
Abstract
Bilateral lower extremity weakness and swelling can have several causes. Although often underdiagnosed, mitochondrial myopathy is more prevalent in the general population than more commonly suspected diseases, such as Guillain-Barre syndrome. The clinical manifestations of mitochondrial disease can be broadly classified into 3 categories: chronic progressive external ophthalmoplegia, skeletal muscle-central nervous system syndromes, or pure myopathy. Cardiac abnormalities occur in 30% to 32% of cases, mostly in the form of hypertrophic cardiomyopathy, dilated cardiomyopathy, or conduction abnormalities. We report a case of a 21-year-old student who developed bilateral lower limb weakness, pain, and swelling diagnosed with mitochondrial myopathy on muscle biopsy. Initial laboratory tests revealed elevated creatinine kinase, brain natriuretic peptide, troponin, myoglobin, and lactic acid and reduced serum bicarbonate. Cardiac workup revealed systolic heart failure with a reduced ejection fraction. Endomyocardial biopsy revealed punctate foci of lymphocytic myocarditis. However, cardiac magnetic resonance imaging did not reveal either myocarditis or an infiltrative cardiac disease. An extensive autoimmune and infection work-up was negative. A muscle biopsy from the patient’s rectus femoris revealed scattered ragged-blue fibers (stained with NADH dehydrogenase), scattered ragged-red fibers on modified Gomori trichrome stain, and cytochrome-c oxidase negative fibers with increased perimysial and endomysial connective tissue, consistent with active and chronic primary mitochondrial myopathy. The patient was treated successfully with furosemide, metoprolol, and methylprednisolone. Adult-onset mitochondrial myopathy is a rare clinical disorder, and our experience stresses the importance of using an inter-disciplinary team approach to diagnose uncommon clinical disorders with widely variable multisystem involvement.
Introduction
Patients with bilateral lower limb weakness are usually evaluated for several disorders, including, but not limited to, stroke, peripheral neuropathy, Guillain-Barre syndrome, multiple sclerosis, disk herniation, etc. The diagnosis of these disorders may not be straightforward when imaging fails to reveal an obvious cause. Mitochondrial myopathies occur in 1:5000 individuals in the general population of the United States and have a delayed clinical onset in 1:10 000 adults. 1 In comparison, Guillain-Barre syndrome occurs in 1 to 2 out of 100 000. 2 Mitochondrial diseases can be easily missed and underdiagnosed, particularly in adults, since these diseases do not have sensitive and specific biomarkers and often require invasive testing to establish a diagnosis. 3 Clinical manifestations can be broadly classified into 3 categories: chronic progressive external ophthalmoplegia, skeletal muscle-central nervous system syndromes (stroke-like episodes), and pure myopathy. Cardiac abnormalities occur in 30% to 32% of cases, mostly in the form of hypertrophic cardiomyopathy, dilated cardiomyopathy, or conduction abnormalities. 1 The clinically recognizable mitochondrial syndromes that can present in the adult population include NARP (neuropathy, ataxia, retinitis pigmentosa), mitochondrial neurogastrointestinal encephalopathy, Kearns-Sayre syndrome, and maternally inherited diabetes and deafness (Table 1). 4
Mitochondrial Disorders.

Source: Mattman et al. 4 Mitochondrial disease clinical manifestations: An overview. BC Med J 2011; 53: 183-187. National Organization of Rare Diseases: raredisease.org.
Case
A 21-year-old male graduate student was referred to our emergency center from the university’s student health center for suspected deep vein thrombosis. He complained of a 3-week history of weakness, pain, and swelling in both legs which began after he arrived in the United States from India. He mentioned “flu-like” symptoms 4 months prior to presentation, which he had assumed was due to a coronavirus (COVID) infection. However, he denied ever undergoing COVID testing. He had received the Covaxin vaccine (a whole inactivated virus vaccine) in India approximately 2 months prior to presentation. He denied similar episodes in the past and had no relevant past medical history or surgical history. His review of systems was negative for decreased visual acuity, hearing loss, and dysphagia. His family history was negative for muscle disorders and ophthalmologic disorders.
On examination, he had bilateral ptosis (which he claims he has had since childhood), limited extraocular movements, tachycardia, jugular venous distension, bilateral 2+ pitting edema up to his knees, bilateral 4/5 Medical Research Council grade weakness of his lower extremities, mild tenderness of his upper and lower extremity proximal and distal muscle groups, and absent deep tendon reflexes with bilateral foot drop. The neurologic consultant determined that the proximal muscle weakness with greater than distal muscle weakness.
His laboratory findings on presentation include a creatinine kinase of 691 IU/l (26-308), brain natriuretic peptide of 3437 pg/ml (<124), troponin of 47.1 ng/l (<19), myoglobin of 195 ng/ml (28-72), lactic acid of 7.7 mmol/l (0.5-2.2), and decreased serum bicarbonate of 12 mmol/l (20-30). His cerebrospinal fluid (CSF) had an elevated protein level (204 mg/dl) but the spinal tap was traumatic (RBC 213 000/mm3, WBC 116/mm3, ratio 1836) which limited the interpretation of the results.
An electrocardiogram showed normal rate and sinus rhythm with left axis deviation and left anterior fascicular block. Chest X-ray and computed tomography (CT) angiography of the chest/abdomen/pelvis showed findings suggestive of volume overload with cardiomegaly, small bilateral pleural effusions, distended inferior vena cava and iliac veins, and body wall edema (Figure 1). His bedside echocardiogram revealed mild left global hypokinesis, a reduced ejection fraction of 40% to 44%, and pulmonary hypertension (RSVP-43 mmHg). He was admitted from the emergency center for further workup and management of his new-onset systolic heart failure with reduced ejection fraction. The primary team concluded that his illness was secondary to an infection. Extensive laboratory testing was negative with the exception of reactive parvovirus IgG, Epstein-Barr IgG, and hepatitis A IgG antibodies. All serologic tests for Coxsackievirus (1-6) were negative. The PCR for herpes 6 DNA revealed <500 copies per ml, a negative result. Autoimmune workup, including ANA, RF, Scl 70, was also negative. The ophthalmology consultant confirmed ophthalmoparesis and ruled out cranial nerve palsy, myasthenia gravis, and retinitis pigmentosa. Gq1b antibody was negative (ruling out the Miller Fisher variant of Guillain-Barre syndrome). Due to decreasing maximal inspiratory pressures and concern about impending respiratory failure, the patient was transferred to the medical intensive care unit.
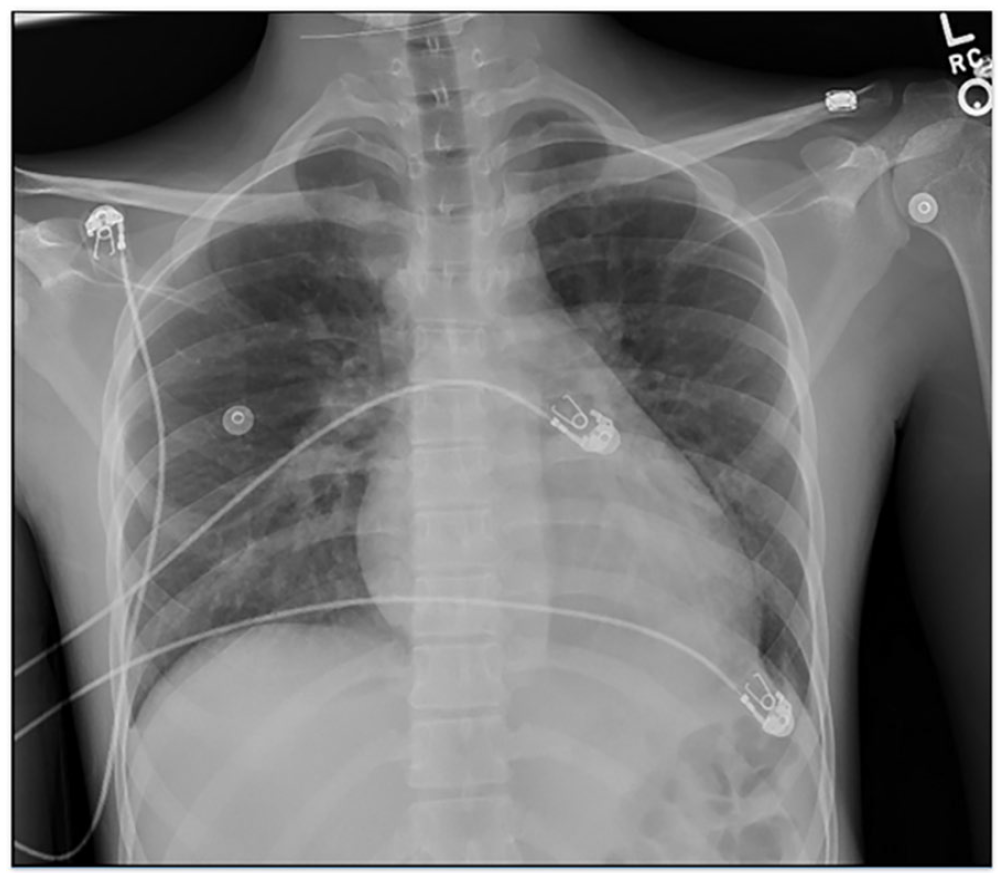
Portable chest X-ray showing cardiomegaly and mild pulmonary congestion.
Mitochondrial myopathy was in the differential diagnosis, and a surgical biopsy of the patient’s rectus femoris revealed scattered ragged-blue fibers, scattered ragged-red fibers, and cytochrome-c oxidase negative fibers with increased perimysial and endomysial connective tissue, consistent with active and chronic primary mitochondrial myopathy (Figures 2-4). An endomyocardial biopsy later revealed punctate foci of active lymphocytic myocarditis with a background of minimal muscle hypertrophy. Magnetic resonance imaging of the heart did not reveal myocarditis or infiltrative changes in the myocardium; the RV ejection fraction was 39% and the LV ejection fraction was 49%.

Rectus femoris muscle biopsy histopathology: muscle fibers failing to stain with cytochrome c oxidase (20×).

Rectus femoris muscle biopsy histopathology: ragged-blue fibers diffusely reactive to succinate dehydrogenase staining (40×).

Rectus femoris muscle biopsy histopathology: ragged-red fibers on modified Gomori trichrome stain (40×).
Genetic analysis by a reference laboratory using next generation sequencing at the Mayo Clinic in Rochester, Minnesota revealed a mitochondrial deletion of m.8482_13447, nearly identical to known deletion m.8470_13446 seen in patients with Kearns-Sayre syndrome. The heteroplasmy level of this deletion was approximately 75% to 80%. This analysis also identified 2 mitochondrial nuclear gene mutations: (1). p. R136C, c.406C > T, chromosome 11, a mutation which results in mitochondrial complex 1 deficiency, if present as an autosomal recessive; (2). p. S122 G, c.364A > G, chromosome 6, a mutation which results in 3-methylglutaconic aciduria and an association with deafness, encephalopathy and Leigh-like syndrome, if present as an autosomal recessive. Both mutations were considered variables of uncertain significance.
The patient was treated successfully with furosemide, metoprolol, and methylprednisolone. On follow up appointments, he reported improved symptoms and was near his baseline. He will require serial electrocardiograms and close follow-up with the cardiology outpatient clinic for the possibility of developing an atrioventricular block and may need a permanent pacemaker in the future.
Discussion
Since the majority of mitochondrial diseases present in childhood, mitochondrial myopathies in adults are likely underdiagnosed, especially in the absence of a strong clinical suspicion. For example, Galán et al 5 described a case of mitochondrial myopathy in a patient who had been misdiagnosed with chronic fatigue syndrome and treated for that for several years. The key findings in our patient that were essential to reach the correct diagnosis were his history of childhood ptosis, muscle tenderness on examination, and elevated lactate and creatinine kinase on presentation. Elevated lactate levels are reported to have a sensitivity ranging from 34% to 62% and a specificity of 83% to 100% for mitochondrial disease. 3 In this case, we could not use the lumbar puncture results to confirm the diagnosis, since elevated CSF protein levels can occur in Kearns-Sayre syndrome as well as GBS. An inter-disciplinary team approach is crucial to evaluating various clinical phenotypes in these patients and ruling out other potential causes.
Mitochondrial Disorders
Determining the frequency of mitochondrial disorders requires clinical analysis to determine phenotypes, genetic analysis to determine mutations, and family studies to determine inheritance. The studies largely occur in a few specialized centers. Schafer and colleagues studied the prevalence of mitochondrial DNA disease in adults in North East England and determined that 9.2 per 100 000 people have a clinically apparent mitochondrial DNA disease. Most of the abnormal genotypes were point mutations; m3242A > G was the most common. 6 In Finland, researchers have focused their study on the MELAS (mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes) syndrome and found a prevalence of approximately 16. 3/100 000 in the adult population (95% confidence interval 11.3-21.4/100 000). 7 Both these studies were done in regions that might be considered as “genetic clusters” of people with similar genotypic composition; the researchers demonstrated that mitochondrial disease is 1 of the most prevalent neurogenetic diseases in adults. 8
Mitochondrial DNA (mtDNA) is a circular structure that has 16 569 base pairs and codes for 13 proteins in the oxidative phosphorylation system, 22 transfer RNAs, and 2 ribosomal RNAs. It consists of 2 strands designated as the heavy strand and the light strand. The number of mitochondria ranges from 100 to 1000 per cell, and each contains 2 to 10 copies of DNA. Eighty percent of the genetic mutations involving the mitochondrial disorders involve mitochondrial DNA; 20% involve nuclear DNA. The types of mutations include point mutations, deletions, and duplications. Nuclear DNA provides some of the enzymes necessary for mitochondrial DNA replication, organization, and maintenance. The number of genetic mutations can occur uniformly throughout a cell; this is referred to as homoplasmy. The number of genetic mutations can vary significantly in the mitochondria in a single cell, between cells in a single tissue, and between tissues; this is referred to as heteroplasmy. These mutations interfere with the production of ATP and can increase the concentration of reactive oxygen species in mitochondria and cells. This results in reduced cellular function secondary to deficiency of energy and increased cellular injury secondary to high levels of reactive oxygen species. This can lead to multiple phenotypes.
A common misconception is that mitochondrial disease occurs only with maternal transmission. Chronic progressive external ophthalmoplegia, Kearns-Sayre syndrome, and Pearson syndrome are associated with variable heteroplasmy of a single mtDNA deletion. These diseases more commonly present as single sporadic somatic events that occurs in the maternal oocyte during early embryonic development rather than germline mutations.9,10 The typical deletion involves 4977 nucleotides. Autosomal recessive, autosomal dominant, and X-linked nuclear genetic disorders occur frequently in adults and can become clinically manifest at any age. 11 Larger deletions are associated with more severe disease and earlier onset. 11
Mitochondrial Syndromes
Kearns-Sayre syndrome, sporadic progressive external ophthalmoplegia, and Pearson’s syndrome are 3 diseases caused by large-scale mitochondrial DNA rearrangements, including partial deletions, and/or partial duplication. Patients with Kearns-Sayre syndrome usually present before 20 years of age. This syndrome is characterized by retinitis pigmentosa (with preserved vision), chronic progressive external ophthalmoplegia (with ptosis being the most common complaint), cardiomyopathy, and elevated CSF protein. Less common clinical findings include cerebellar symptoms (ataxia), weakness of muscles of the face, pharynx, trunk, or extremities, and progressive hearing loss. A skeletal muscle biopsy in an individual with Kearns-Sayre syndrome would show ragged red fibers and abnormal mitochondria. 12 These patients develop electron transport chain complex I/IV deficiencies. In a study measuring the biochemical changes in Kearns-Sayre syndrome, nicotinamide adenine dinucleotide (NAD)-linked substrates and succinate were much lower than those of control subjects, and cytochrome oxidase activity was decreased. 13
Khambatta et al 14 published a case series of 35 patients with Kearns-Sayre syndrome from the Mayo Clinic in 2014. The mean age at diagnosis was 26 years; the mean follow-up period was 10.8 years. None of the patients had a family history of Kearns-Sayre syndrome. Based on information collected during this follow-up period, 31 patients (88.6%) had external ophthalmoplegia, 30 patients (85.7%) had ptosis, and 25 patients (71.4%) had pigmentary retinopathy.About 27 patients (77.1%) had weakness, 16 patients (45.7%) had ataxia, 11 patients (31.4%) had dysphagia, and 14 patients (40.0%) had deafness. About 6 patients (17.1%) had a dilated cardiomyopathy, 11 patients (31.4%) had heart block on ECG, and 23 patients (65.7%) had conduction delays. About 6 patients (33.3% out of 18 tested) had elevated lactate levels. Information collected from this relatively large cohort of patients indicates that these patients do not have a uniform clinical presentation and that some do have cardiac disease based on echocardiography and electrocardiograms.
Limongelli et al 15 also studied the natural history of adult patients with mitochondrial disorders and heart disease. This study included 32 patients with a mean age of 37.8 years. These patients had ECGs, 24-h ECG monitoring, and cardiopulmonary exercise testing. About 22 patients (68%) had had abnormal ECGs, and 19 patients (44%) had abnormal echocardiograms. About 23 patients completed a metabolic exercise test; 6 patients stopped with dyspnea, and 17 patients stopped with muscle fatigue. During follow-up over a mean period of 4.1 years, 4 patients (12.5%) developed arrhythmias or syncope requiring device therapy, and 1 patient required heart transplantation.
Kabunga et al 16 and co-authors published a detailed systematic review of cardiac conduction disorders in patients with Kearns-Sayre syndrome. About 50% of these patients develop cardiac complications. The most frequent cardiac disorder was conduction system disease which could progress to complete atrioventricular block or bradycardia-related polymorphic ventricular tachycardia. These authors suggested that these patients should have regular ECGs to identify conduction system disease which has the potential to develop bradycardia and malignant tachyarrhythmias. Finsterer 17 summarized the comprehensive management needed in patients with Kearns-Sayre syndrome who have cardiac disease. He notes that these patients are at risk for sudden cardiac death and may need pacemaker implantation and eventually implantable cardioverter defibrillators.
Case Details
The patient in this case report had multiple features typically seen in patients with Kearns-Sayre syndrome. At presentation, he had significant cardiac and skeletal muscle injury. He did not have retinitis pigmentosa, hearing loss, or dysphagia. The genetic analysis indicates that he had a large deletion in his mitochondrial DNA. In addition, he had 2 other point mutations in his nuclear mitochondrial DNA. Whether or not these mutations contributed to his presentation is impossible to determine but could be important. The patient had an endomyocardial biopsy which revealed punctate foci of lymphocytic myocarditis. The aggregate volume of these areas was not adequate to be identified by magnetic resonance cardiac imaging. The explanation for these histologic changes is uncertain. They could represent a mild inflammatory response to myocardial apoptosis related to the production of excessive reactive oxygen species in patients with abnormal mitochondrial function. The differential diagnosis of cardiac disease in a young adult would include congenital heart disease, autoimmune disorders such as systemic lupus and polymyositis, and sarcoidosis. However, extensive laboratory testing, radiographic imaging studies, and biopsies from the heart and muscle ruled out these possibilities.
Massively parallel or next-generation sequencing is the preferred method for genetic mutation testing in mitochondrial DNA. It may also be helpful to characterize mtDNA mutations in tissue samples to account for heteroplasmy seen in blood samples and to assess the risk of other organ involvement. Based on consensus recommendations, an open vastus lateralis muscle biopsy should be performed when DNA testing cannot confirm the diagnosis. Analysis of the biopsy tissue should include staining with cyclooxygenase (COX), succinate dehydrogenase (SDH), NADH-tetrazolium reductase, and combined SDH/COX stain along with electron microscopy. 3
Treatment
Multiple treatment modalities have been studied with no widely accepted guidelines for management (Table 2). According to the consensus recommendations from the Mitochondrial Medicine Society, CoQ10 (ubiquinol) should be tried in most patients with mitochondrial myopathies along with alpha-lipoic acid and riboflavin. L-carnitine and other vitamin supplementation are indicated only in deficiency states. 3 These are relatively safe medications which do not have a predictable effect in these patients. However, given the lack of established medical protocols, empiric therapy seems reasonable at least for a period of time. Glover et al 18 randomized 30 patients with mitochondrial disorders to either CoQ10 (1200 mg/day) or placebo for 60 days in a double-blind crossover trial. This drug had minor effects on cycle exercise aerobic capacity and post exercise lactate levels but did not have any effect on other important clinical variables, such as blood lactate, urinary markers of oxidative stress, activities of daily living and quality of life, forearm hand grip strength, and oxygen desaturation. This study indicates that pharmacologic approaches to limit oxidative stress may not provide much benefit in these patients. Other approaches have been tried. For example, 1 case report described improvement in generalized weakness and motor strength in a 20-year-old with adult-onset Leigh syndrome after plasmapheresis and monthly intravenous immunoglobulin infusions. Although this may be attributed to improvement of a concomitant autoimmune process triggered by her pre-existing mitochondrial disease, more research is needed to explore the role of immunotherapy in mitochondrial disorders.19-22
Drugs Used in Mitochondrial Disorders.

Source: Mattman A et al. 4 Diagnosis and management of patients with mitochondrial disease. BC Med J 2011; 53: 177-182.
Barcelos et al 23 published a comprehensive update on medical therapy for mitochondrial disorders in 2020. These authors noted that the main goal of these medications is to stabilize and enhance residual metabolic function(s) in mitochondria to slow clinical disease progression. This treatment often involves combinations of medications, including vitamins, antioxidants, and cofactors. For example, they recommended a B vitamin complex, ubiquinol, and an antioxidant, such as vitamin E, plus folinic acid in patients with Kearns-Sayre syndrome. The main difficulty with empiric trials in these patients is deciding whether or not the medication has any beneficial effects. Formal exercise testing with a 6-min walk test might provide the simplest method to monitor these patients’ clinical course during treatment.
There is no official FDA approved medication for mitochondrial disease, but idebenone (synthetic analog of coenzyme Q10) has been approved for use in treatment of MELAS in Europe. Cellular level interventions, such as gene therapy, augmentation of mitochondrial biogenesis, restoration of the cellular NAD+ to NADH ratio, increasing mitophagy, metabolic reprograming, or manipulation of oxidative stress, hold promise for future advances in the management of mitochondrial disease. 24 Endurance exercise (following cardiac screening) can increase mitochondrial enzyme activity, and high-intensity interval training has been shown to induce similar mitochondrial adaptations, building up conditioning as tolerated by the patient. Patients with mitochondrial disease are at higher risk for general anesthesia related complications, including propofol infusion syndrome. They should wear a Medic Alert bracelet and carry an emergency care plan detailing their disease condition and management recommendations. 3
Patient Care Standards
The Mitochondrial Medicine Society published a consensus statement regarding patient care standards in patients with primary mitochondrial disease in 2017. 25 Their recommendations for managing cardiac disorders in these patients included a standard 12-lead electrocardiogram and echocardiogram at baseline. A 24-h ECG monitor might be considered in patients at high risk for developing cardiac arrhythmias, including patients with cardiomyopathy. Follow-up evaluation should include clinical assessment, blood pressure measurement, ECG, and echocardiograms every 12 months for at least 3 years. Cardiac magnetic resonance images may be needed in some patients with inconclusive cardiac evaluations. Exercise testing can help evaluate functional capacity and responses to medication trials. Clinicians should have a low threshold for pacemaker implantation and possibly implantable defibrillators.
Conclusion
Mitochondrial myopathy is usually not included in the differential diagnoses of adults who present with bilateral lower extremity weakness. Our case demonstrates the need for a systematic approach to interpret the musculoskeletal, neurological, ophthalmological, and cardiac components of these disorders. When neuroimaging is unrevealing, pertinent labs, such as elevated lactate and creatine kinase without an obvious explanation, indicate the need for additional evaluation which should include tests for mitochondrial DNA disorders. Genetic testing is the gold standard for diagnosing mitochondrial disease; an open vastus lateralis biopsy can also be used for confirmation. Recommended treatment trials should include CoQ10, alpha-lipoic acid, and riboflavin supplementation as a trial; endurance exercise following cardiac screening can improve muscle conditioning. If patients undergo endomyocardial biopsy, samples should be referred to specialized laboratories for detailed histologic, biochemical, and electron microscopic study.
Footnotes
Author Contributions
All authors participated in the literature review and case analysis. KB and KN did the initial manuscript drafting.KN revised and finalized the manuscript. All authors approved the final version of the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.