
Abstract
Ethylmalonic encephalopathy is a rare autosomal recessive mitochondrial disorder caused by biallelic pathogenic variants in ETHE1, the gene encoding mitochondrial persulfide dioxygenase, an enzyme crucial for hydrogen sulfide (H2S) detoxification. Loss of this enzyme results in H2S accumulation, cytochrome c oxidase inhibition, oxidative stress, and disrupted energy metabolism. Clinically, ethylmalonic encephalopathy manifests during early infancy with developmental delay, hypotonia, progressive encephalopathy, seizures, chronic diarrhea, and microvascular abnormalities such as petechiae and acrocyanosis. Fewer than 100 cases have been reported globally, mostly among Mediterranean and Arab populations, with scarce data from Latin America. We report the first documented case of ethylmalonic encephalopathy in a Mexican patient. The affected male infant, born to healthy nonconsanguineous parents of indigenous Maya origin from Yucatán, presented at 2 weeks of age with persistent hemorrhagic diarrhea, followed by metabolic acidosis, hyperammonemia, hyperlactatemia, elevated C4-acylcarnitine, and increased urinary ethylmalonic acid. Neurological findings included developmental delay, hypotonia, and myoclonic epilepsy. Whole-exome sequencing revealed a homozygous frameshift pathogenic variant in ETHE1 (NM_014297.5):c.19_20dup (p.Val8Glyfs*7), predicted to introduce a premature stop codon and abolish protein function. Despite targeted interventions—antiepileptic therapy, ammonia-lowering treatment, and metabolic support—the patient’s condition progressively worsened, culminating in death at 15 months after metabolic decompensation and brain death. This case broadens the known mutational spectrum of ETHE1 by identifying a previously unreported pathogenic variant and underscores the need to include ethylmalonic encephalopathy in the differential diagnosis of infants presenting with chronic diarrhea, vascular lesions, and neurological deterioration, even in regions where the condition is not typically observed.
Introduction
Ethylmalonic encephalopathy (EE) is caused by homozygous pathogenic variants in the ETHE1 gene, located on chromosome 19q13. This gene consists of 7 exons and produces a transcript of approximately 1000 nucleotides. It encodes persulfide dioxygenase ETHE1, a mitochondrial protein with a molecular weight of 30 kDa that plays a crucial role in hydrogen sulfide (H2S) catabolism within the mitochondrial matrix. ETHE1 utilizes molecular oxygen to catalyze the oxidation of persulfide after its transfer to a thiophilic acceptor, such as glutathione. The expression of ETHE1 is predominantly localized to the mitochondrial matrix across various tissues, with particularly high activity in the liver, brain, muscle, and intestinal mucosa. Impaired ETHE1 function results in the chronic accumulation of hydrogen sulfide, which is believed to drive most of the biochemical and clinical manifestations of EE. 1
Clinically, patients present with early-onset progressive encephalopathy, global developmental delay, hypotonia evolving into dystonia, spastic tetraparesis, and ultimately, global neurological deterioration. Chronic hemorrhagic diarrhea and petechiae are also prominent findings, as hydrogen sulfide exerts vasoactive and vasotoxic effects, leading to mucosal and vascular endothelial damage. 2
Biochemically, excessive hydrogen sulfide inhibits cytochrome c oxidase and short-chain acyl-CoA dehydrogenase activity, resulting in elevated lactate levels and increased C4/C5 acylcarnitines. 3 Despite these insights, the pathophysiological mechanisms of EE remain incompletely understood. Studies in human and animal models suggest that hydrogen sulfide and ethylmalonic acid accumulation disrupt mitochondrial function and redox homeostasis, leading to oxidative stress. However, emerging evidence indicates that hydrogen sulfide may be the primary driver of toxicity in EE. 4
To date, fewer than 100 cases of EE have been identified worldwide, with most reported cases occurring in individuals of Mediterranean and Arabic descent.2,5–15 However, EE has been rarely documented in Latin American populations. Here, we present the first reported case of EE in a Mexican patient, identified with a homozygous nonsense biallelic pathogenic variant in the ETHE1 gene, predicted to result in a complete loss of ETHE1 expression.
Case report/case presentation
We describe a male patient from Yucatán, Mexico, born to healthy, nonconsanguineous parents, with an unremarkable neonatal course. He was the second child of the couple, and there was no significant family history of genetic or metabolic disorders. The pregnancy was diagnosed during the second trimester, at which point prenatal care and maternal multivitamin supplementation were initiated. Delivery occurred at 38 weeks of gestation via cesarean section due to oligohydramnios, with an APGAR scores of 6 and 9 at 1 and 5 min, respectively.
At 2 weeks of age, the patient developed bloody diarrhea, prompting medical evaluation. Stool examination revealed Entamoeba histolytica cysts, for which metronidazole therapy and a lactose-free formula were prescribed; however, no clinical improvement was observed. Routine neonatal screening performed at 3 weeks of age was reported as normal.
At 7 months of age, the patient was hospitalized due to intermittent diarrhea, dehydration, and malnutrition. Two months later, a panendoscopy and colonoscopy were performed as part of the diagnostic workup. Findings included a nodular pattern in the rectum and sigmoid colon, and multiple aphthoid-type ulcers measuring approximately 3 mm in diameter in the transverse colon. Both regions showed edema, erythema, and loss of vascular pattern, features suggestive of an underlying inflammatory bowel disease, with a suspected diagnosis of Crohn’s disease (Figure 1).

Colonoscopy performed at 9 months of age showing: (a) Rectum and sigmoid colon with a nodular pattern, edema, erythema, friability, and loss of vascular and haustral pattern. (b) Transverse colon with edema, erythema, loss of vascular pattern, and multiple aphthoid-type ulcers measuring approximately 3 mm in diameter.
Additionally, petechiae were observed on both legs and the face, and orthostatic acrocyanosis (Figure 2). During hospitalization, the patient exhibited clusters of spasms that resolved spontaneously, prompting an evaluation by pediatric neurology. Electroencephalography showed irregular background activity with a subclinical epileptic variant, while a brain computed tomography (CT) demonstrated frontal cortical atrophy and bilateral hypodense areas in the corona radiata. Magnetic resonance of the brain showed no pathological findings. Treatment with vigabatrin was initiated, leading to seizure resolution.

Clinical images showing petechiae on the forehead and limbs, and orthostatic acrocyanosis, indicative of microvascular damage in ethylmalonic encephalopathy.
Given the combination of neurological and gastrointestinal symptoms, an inborn error of metabolism was suspected. Biochemical testing demonstrated hyperglycemia (glucose: 142.75 ± 50.8 mg/dL), marked hyperammonemia (125.32 ± 76.44 µmol/L), increased urinary excretion of ethylmalonic acid, metabolic acidosis (pH: 7.27 ± 0.17; bicarbonate: 10.17 ± 3.33 mmol/L), and hyperlactatemia (9.63 ± 5.51 mmol/L). Evidence of hepatic dysfunction was also observed, with elevated alkaline phosphatase (103.78 ± 27.5 U/L) and lactate dehydrogenase (995.92 ± 956.99 U/L). Expanded metabolic screening revealed elevated C4-acylcarnitine levels (mean ± standard deviation: 2.17 ± 0.33 µmol/L).
In the differential diagnosis for elevated C4-acylcarnitine, multiple acyl-CoA dehydrogenase deficiency was initially considered but ruled out based on the presence of ethylmalonic aciduria. The biochemical profile characterized by elevated C4-acylcarnitine and ethylmalonic aciduria narrowed the differential to short-chain acyl-CoA dehydrogenase deficiency and EE. To confirm the diagnosis, whole exome sequencing was performed, revealing a biallelic pathogenic variant in ETHE1 (NM_014297.5):c.19_20dup(p.Val8Glyfs*7), confirming the diagnosis of EE.
The patient was discharged with a management plan guided by current evidence-based recommendations, 16 including metronidazole (15–30 mg/kg/day) to lower urinary ethylmalonic acid and reduce H2S levels, levetiracetam and vigabatrin for seizure control, and sodium bicarbonate to correct metabolic acidosis. Regular follow-up, genetic counseling and biochemical monitoring were implemented to monitor the patient’s condition and optimize the management of EE. Serial blood ammonia showed a significant improvement with treatment, decreasing from an initial level of 257 µmol/L at diagnosis to 60 µmol/L during follow-up.
At 1 year and 3 months of age, the patient experienced rapid clinical deterioration and was admitted to the pediatric emergency department with diarrhea, complicated with hypovolemic shock, compensated metabolic acidosis, and respiratory failure, requiring assisted mechanical ventilation. Despite intensive management, hospital-acquired pneumonia developed. Forty-eight hours later, the patient exhibited bilateral mydriasis with fixed, nonreactive pupils; brain CT imaging revealed bilateral hypodensities consistent with brain death. Progressive hemodynamic deterioration ensued, culminating in asystole.
Genetic findings
WES revealed an homozygous variant in ETHE1(NM_014297.5):c.19_20dup(p.Val8Glyfs*7), which is a frameshift variant that introduces a premature translational stop codon in position 7 of the protein (p.Val8Glyfs7), likely resulting in an absent or nonfunctional protein product. According to the American College of Medical Genetics and Genomics (ACMG) classification, this variant is considered pathogenic, meeting the following criteria: PVS1, as it is a null variant in a gene where loss of function is a well-established disease mechanism; PM2, due to its extremely low frequency in gnomAD population databases; PP4, patient’s phenotype is highly specific for a disease with a single-genetic etiology and PP5, based on its classification as pathogenic by a reputable source. To the best of our knowledge, this variant has not been previously reported in the literature in association with ETHE1-related conditions.
Discussion
EE is an inherited metabolic disorder resulting from biallelic pathogenic variants in the ETHE1 gene, which encodes the mitochondrial enzyme persulfide dioxygenase. This enzyme oxidizes persulfides after their transfer to a thiophilic acceptors such as glutathione, thereby contributing to mitochondrial metabolic homeostasis. Loss of ETHE1 function leads to the accumulation of H2S, which inhibits cytochrome c oxidase activity. Therefore, affected individuals exhibit elevated levels of lactate, C4 and C5-acylcarnitines in plasma, along with increased urinary ethylmalonic acid 1 —hallmark biochemical findings of the disease.
This case describes a patient with EE caused by truncating homozygotic variants in ETHE1. Both parents were of Mexican origin and belonged to an indigenous Maya community. Absence of consanguinity contrasts with most reported cases, which predominantly involve Mediterranean or Arab families with consanguineous unions.7,8 Laboratory findings in our patient, including elevated C4-acylcarnitine, hyperlactatemia, metabolic acidosis, and hyperammonemia, are consistent with the underlying pathophysiology of H2S accumulation and inhibition of cytochrome c oxidase and short-chain acyl-CoA dehydrogenase. 1
Clinically, the patient initially presented with bloody diarrhea at 2 weeks of age, which remained the only symptom until 9 months of age, when cluster spasms were observed. This neurological manifestation prompted electroencephalographic evaluation, revealing a pattern consistent with myoclonic epilepsy. The early presentation limited to gastrointestinal symptoms made it challenging to suspect an inborn error of metabolism, as such findings are more commonly attributed to higher-prevalence disorders, including celiac disease or irritable bowel syndrome. Moreover, the endoscopic findings resembling those of inflammatory bowel disease led to an initial suspicion of Crohn’s disease, which can mimic the gastrointestinal phenotype of EE. However, the subsequent emergence of neurological manifestations in association with the diarrheal syndrome redirected the diagnostic approach toward a metabolic etiology.
This case underscores the importance of maintaining a high index of suspicion for metabolic diseases in infants presenting with persistent diarrhea, particularly when accompanied by systemic manifestations such as developmental delay, hypotonia, seizures, or microvascular signs including petechiae and acrocyanosis. Early recognition of such disorders is crucial for guiding appropriate diagnostic testing and timely therapeutic intervention. 17
The patient passed away due to metabolic decompensation triggered by multiple infectious processes, an outcome widely reported in the literature, as intercurrent infections are known to precipitate significant clinical deterioration.7,17 In contrast, individuals with milder phenotypes may exhibit a more favorable prognosis, maintaining adequate neurodevelopment during childhood and remaining free of complications such as seizures, developmental regression, or acrocyanosis. 13 Furthermore, survival into adolescence has been documented, although persistent manifestations such as chronic diarrhea, neuromotor delay, and acrocyanosis are often present. 15
In the present case, the severe clinical course and early fatal outcome may be attributable to recurrent infectious leading to metabolic decompensation, compounded by the underlying genetic defect. The identified ETHE1 variants introduce a frameshift predicted to result in complete loss of protein function. This finding aligns with the observed phenotype severity. In contrast, other reports have described patients carrying missense variants associated with milder disease progression and longer survival, as illustrated by the cases reported by Platt et al. 15 As summarized in Table 1, these patients exhibited extended survival and distinct clinical profiles compared to our case, likely because missense variants may permit the production of a partially functional enzyme, albeit at reduced efficiency. 1
Clinical features of the patient compared with published cases reported by Platt et al. 15 .
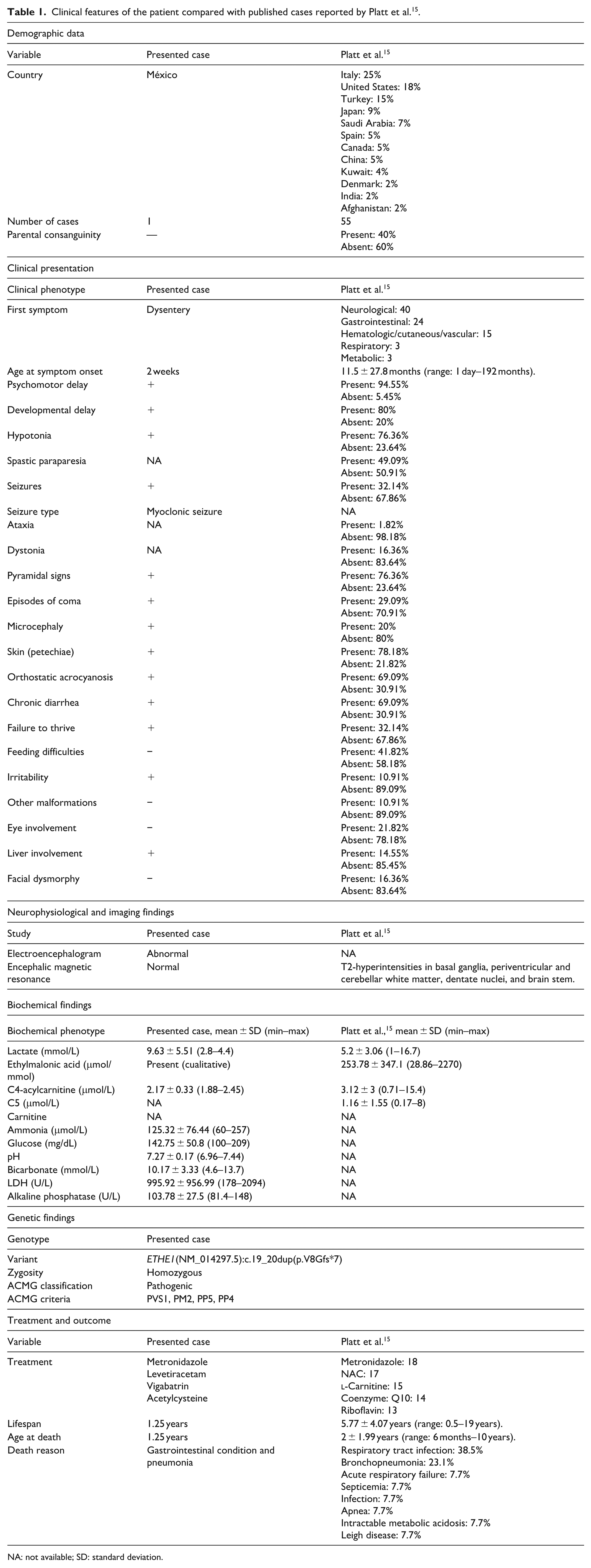
NA: not available; SD: standard deviation.
The definitive diagnosis of EE relies on molecular confirmation of pathogenic variants in ETHE1. Various pathogenic changes have been identified in the ETHE1 gene, including nonsense and missense mutations, as well as deletions affecting single or multiple exons. Notably, deletions involving exon 4 or the entire gene appear to be the most frequent alterations reported. 17 The variant ETHE1(NM_014297.5);c.19_20dup identified in this case, represents a novel finding. This frameshift mutation introduces a premature stop codon, likely resulting in a truncated, nonfunctional protein and explaining the sever phenotype observed.
This case expands the molecular and geographic spectrum of EE by documenting the first report of a truncating ETHE1 variant in an individual of Mexican and indigenous Maya origin. The clinical course underscores the diagnostic challenges posed by early gastrointestinal manifestations and highlights the importance of considering metabolic disorders in infants presenting with unexplained chronic diarrhea and subsequent neurological decline. Early recognition and molecular diagnosis are essential for genetic counseling, prognostic assessment, and potential implementation of emerging therapeutic strategies aimed at mitigating disease progression and improving survival.
Conclusion
We report a patient carrying a novel ETHE1 variant ([NM_014297.5]:c.19_20dup) who presented with elevated blood levels of C4-acylcarnitine, ammonia, and lactate, together with clinical features characteristic of EE, including psychomotor delay, hypotonia, microvascular involvement, chronic diarrhea, and seizures. To our knowledge, this is the first description of this specific variant and the first reported case of EE in an individual of Mexican origin.
Footnotes
Ethical considerations
Ethical approval to report this case was obtained from the Research and Ethics committee of The General Hospital “Dr. Agustin O’Horan,” approval number CI-015-1-25.
Consent to participate
Written informed consent was obtained from a legally authorized representative for anonymized patient information to be published in this article.
Author contributions
DARM: Clinical analysis of the case, literature review, writing the initial draft of the manuscript, revising the manuscript critically.
ERVP: Clinical analysis of the case, literature review, writing the initial draft of the manuscript, revising the manuscript critically.
YQP: Medical management of the case, revising, and approving the final draft, revising the manuscript critically.
HLM: Medical management of the case, revising, and approving the final draft, revising the manuscript critically.
CAMC: Medical management of the case, revising, and approving the final draft, revising the manuscript critically.
FJCG: Medical management of the case, literature review, writing the initial draft of the manuscript, conducted genetic and bioinformatic studies, revising, and approving the final draft, revising the manuscript critically.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: CONAHCYT—Beca Nacional—Funding Number: 2022-000002-01NACF-00427
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
All data generated or analyzed during this study are included in this article. Further inquiries can be directed to the corresponding author.