
Abstract
Elevated lipid panels are associated with an increased risk of cardiovascular disease. Management of heart disease with lipid lowering agents play a vital role in medicine. Statins are one group of medications that are widely utilized in the medical field to decrease the risk of atherosclerotic disease. Statins work by inhibiting the hepatic enzyme 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR). Although statins are one of the most effective drugs for secondary and primary prevention of heart disease, they are not without risks and side effects such as hepatotoxicity and myopathy. We present a case of a male patient who developed progressively worsening muscle weakness and elevated muscle enzyme markers upon initiation of a statin. His symptoms persisted despite a trial of an alternative statin and subsequent discontinuation of all statin medications. A multitude of possible etiologies were considered and ranged from infectious, autoimmune, cancerous, to congenital in nature. Environmental factors, such as exposure to medications or toxins, were also considered as one of the possible precipitating factors. The association between his statin consumption and muscle weakness were not easily apparent at first. He required further workup including physical examination, electromyography, panel of myositis antibodies, and muscle biopsy. After clinical suspicion and elevated antibodies to HMGCR beyond the normal limit, he was discovered to have statin-associated autoimmune myopathy. The patient improved with the treatment of immunosuppressive agent’s prednisone and methotrexate.
Keywords
Case Presentation
A 74-year-old male with a past medical history of glaucoma, tension-type headaches, and coronary artery disease presented to our facility for evaluation of progressively worsening bilateral lower extremity weakness. His lower extremity muscle weakness first began 5 years ago after he underwent a multivessel coronary artery bypass graft (CABG). After the CABG, he started atorvastatin 80 mg for secondary prevention of atherosclerotic cardiovascular disease.
Upon initiation of the statin medication, he noticed a gradual increase of weakness to complete tasks. By the time of his presentation to our facility, his muscle weakness had progressed to the point where he could not cross his legs. He lacked any history of congenital muscular defects, traumatic injury, orthopedic or neurosurgical procedures. The patient never had any prior exposure to steroids or toxins. His personal and family history were negative for muscular disorders, including mitochondrial diseases, muscular dystrophy, or autoimmune conditions.
Musculoskeletal examination demonstrated weakened strength in his quadricep and hip flexor muscles of 2 out of 5 in severity. There was noticeable symmetrical atrophy over the triceps, deltoid, biceps and bilateral proximal shoulder girdles also consistent with a strength of 2 out of 5 in severity. Gower’s sign was negative. There were no signs of ataxia or bradykinesia upon assessment of his gait. His deep tendon reflexes were intact and adequately evoked at the biceps, triceps, brachioradialis, knees, and ankles bilaterally. Sensation to light touch and pain were intact in all extremities. The patient lacked any dermatological skin findings that would suggest an inflammatory muscle disorder. All other aspects of his physical exam were unremarkable including cardiopulmonary and abdominal examination.
Laboratory workup illustrated normal inflammatory markers of C-reactive protein (CRP) and erythrocyte sedimentation rate (ESR). His serum creatinine kinase (CK) level averaged around 99 U/L prior to his statin initiation. After he first started atorvastatin, his CK became markedly increased, peaking at 5700 U/L. The dose of atorvastatin was decreased and although his CK had improved it still remained elevated at around 4162 U/L. Additionally his serum aldolase level ranged around 77.6 U/dL. Further laboratory work up including levels of aldolase, creatine kinase, aspartate transaminase, and alanine transaminase were attained and monitored throughout his clinical course. His liver function enzymes were mildly elevated but not concerning for hepatotoxicity given that they both averaged the low 60s U/L.
Once he was weaned off the atorvastatin, his weakness symptoms slightly improved but persisted. Additionally, after weaning off the statin his CK levels improved steadily over the next several months. Almost 6 months after the discontinuation of atorvastatin, he was placed on a trial of rosuvastatin but this also led to a gradual increase in his CK that peaked at a level of 3393 U/L (Figure 1).
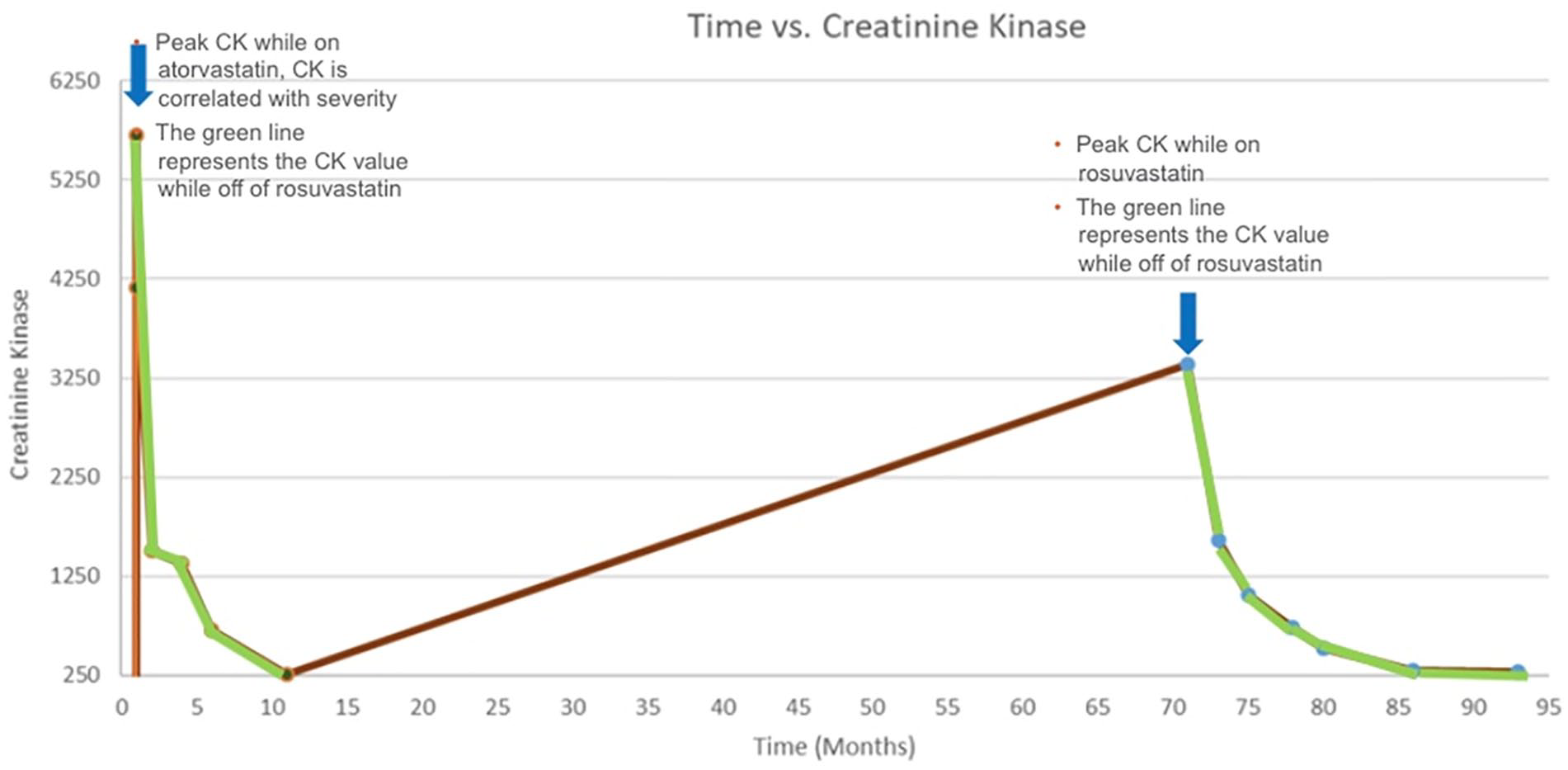
Time in months versus creatinine kinase. This graph represents the patients CK level (muscle enzyme, correlated with severity of myopathy) over time in months. The graph plots the values of the CK while on atorvastatin, rosuvastatin, and while off all statins (green line) over time.
The patient underwent serum laboratory workup that included an autoimmune antibody panel, complement, hepatitis and HIV work up and all these results were negative. The patient also underwent an extensive myositis-specific autoantibodies (MSAs) panel (Table 1).
The Results of Serum Myositis Antibody Panel.
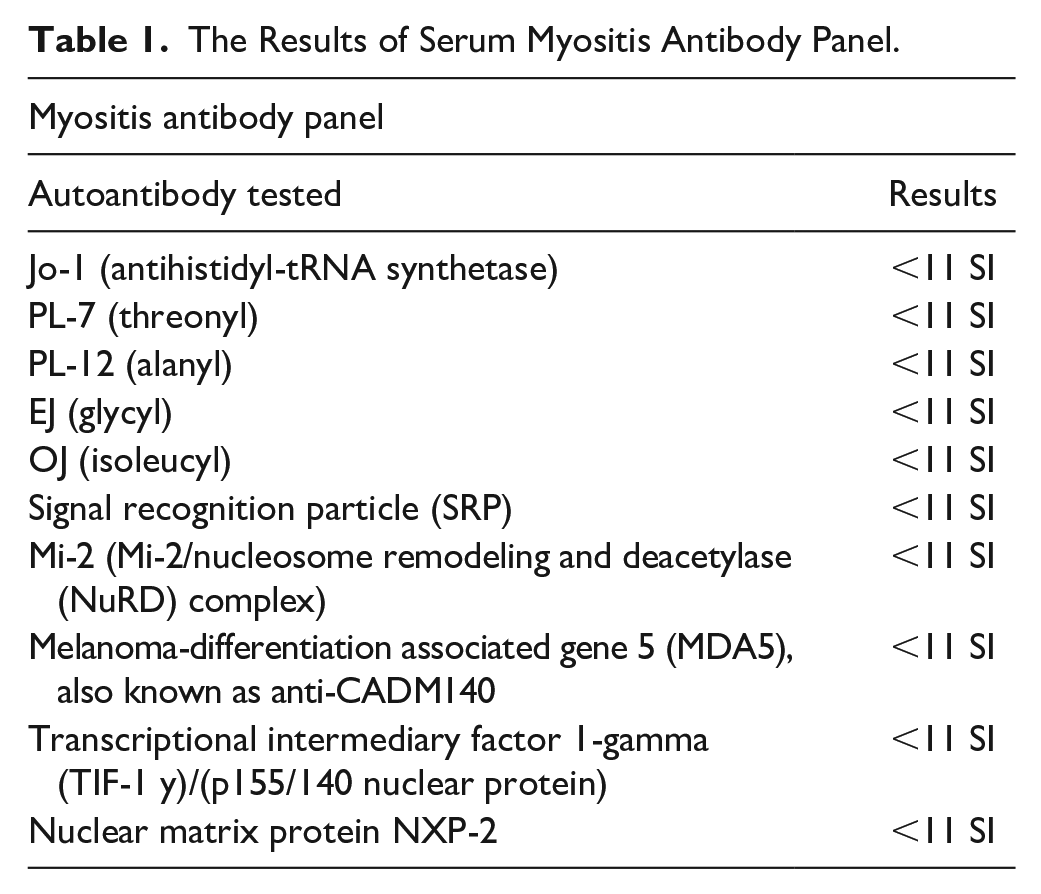
The results of his comprehensive full myositis antibody panel are presented in this table; all of the results were found to be negative. Quest Diagnostics performed this test. The first 5 autoantibodies in the table include Jo-1 (antihistidyl-tRNA synthetase), PL-7 (threonyl), PL-12 (alanyl), EJ (glycyl), and OJ (isoleucyl); these are myositis specific autoantibodies that pertain to cytoplasmic enzyme reactions involving tRNA and specific amino acids. Signal recognition particle (SRP) is another set of myositis antibodies that were found to be negative. Mi-2 and NuRD autoantibodies tend to be positive in patients with dermatomyositis. The autoantibody to the protein of (MDA5)/anti-CADM 140 is typically positive in patients with amyopathic dermatomyositis (CADM). Autoantibodies to (TIF-1 y) are normally found in adult and juvenile dermatomyositis. Autoantibodies to nuclear matrix protein NXP-2 are associated with juvenile dermatomyositis (JDM).
The myositis antibody panel included autoantibodies to proteins and enzymes that may lead to myositis, but the results were normal and therefore the patient underwent an electromyographic (EMG) diagnostic procedure for further evaluation. Using the monopolar electrode technique, EMG examination was carried out on the main muscles of the left lower and upper extremity. In all of the muscles studied, insertional activity was normal. There was no evidence of positive sharp waves, fibrillation potentials, or repetitive discharges. During voluntary contraction, the motor units seen were of normal amplitude and duration. Recruitment was normal and there was no electrophysiologic evidence of acute myopathic or neuropathic process affecting the upper or lower extremities.
The EMG was normal; therefore, a decision was made to perform muscle biopsy of the left thigh with analysis of a 2 cm sample for further information (Figure 2).

Pathology results of the left anterior thigh muscle biopsy: (a) This is an H&E stain of a degenerating muscle fiber. The arrow points to degenerating myofiber. Stars indicate increased internal nuclei which is a nonspecific myopathic feature. There are also myofibers of various sizes and shapes; this is also a nonspecific myopathic feature, (b) CD68 showing it is degenerating by macrophages. CD3 stains there are minimal. T-lymphocytes and those present are associated with degenerating fibers or in endomysial space. There are no T-cells invading viable myofibers. Skeletal muscle with myopathic features including myofiber degeneration and regeneration. This skeletal muscle biopsy reveals myopathic features with evidence of scattered myofiber degeneration and regeneration and foci of endomysial macrophage infiltration associated with areas of myofiber necrosis.
Upon serological testing, the patient had anti-HMGCR antibodies at a level of 200 CU (reference range 0-19 units: Negative). Based upon the correlation of his statin use and symptoms, elevated muscle enzymes and the seropositivity of anti-HMGCR antibodies a diagnosis of statin-associated autoimmune myopathy was formulated. Upon the diagnosis, all statin medications were discontinued indefinitely. He eventually started evolocumab injections for further management of hyperlipidemia and secondary prevention of cardiovascular disease. The patient also started disease-modifying therapy methotrexate 17.5 mg with low-dose prednisone 10 mg per day and tolerated both medications well. The patient responded immediately within the first few months of these 2 medications and illustrated improvement over time. He exhibited complete recovery within a year to the point where he was able to return to his daily activities.
Discussion
In 2004, the European Neuromuscular Center (ENMC) added immune-mediated necrotizing myopathy (IMNM) as a subtype of idiopathic inflammatory myopathy (IIM). 1 IMNM is characterized by symmetrical proximal weakness with myofiber necrosis and minimal cellular inflammatory infiltration on muscle biopsy. 1 The ENMC updated the IMNM diagnostic criteria in 2016 to include myositis-specific antibody (MSA) profiles. Although IMNM cannot be ruled out in seronegative individuals, patients with anti-signal recognition particles (SRP) or (HMGCR) antibodies can be identified with IMNM. 2 We present a case of anti-HMGCR immune mediated necrotizing myopathy induced by statin.
The possible mechanism by which statins trigger the onset of IMNM is due to upregulation of HMGCR expression, and depending on certain immunogenetic parameters, the body reacts by triggering an autoimmune response. It is noted that the regenerating muscle fibers express the HMGCR protein in large quantities. With exposure to statins, the result is a consistently high concentration of this protein and a disruption of the balance between cell destruction and repair.1,2 Our patient was using atorvastatin and according to the latest data, this drug class is strongly associated with the development of anti-HMGCR positive antibody and IMNM compared with simvastatin or rosuvastatin. 3 The incidence of this entity is estimated at 1/1 000 000 per year with female predominance over 40 years of age. 4
The clinical manifestations are progressive subacute proximal symmetrical muscle weakness, which is key to the clinical diagnosis. Additionally, limb weakness, dysphagia, and myalgia are found more commonly in patients with statin-induced-myopathy as compared to patients with dermatomyositis and polymyositis.5,3 The CK level is often elevated to levels of 6000 U/L or more. 5 Our patient had a CK of 5700 U/L on admission. However, a case reported by Kang et al 7 on a 68-year-old male patient who had mild weakness with a CK level not exceeding 657 U/L. Recent research has identified statin-induced IMNM in patients with increased CK but normal muscular strength. 4
The mean duration between onset of symptoms and start of statins is highly variable ranging from 2 months up to 10 years. 6 Our patient was diagnosed after 5 years of onset of symptoms. This means that thinking of statin-induced myotoxicity only when the duration is short is a misconception that can delay diagnosis and therefore management. Thus, the clinician must have a high clinical suspicion to consider this condition as a possible differential diagnosis. Moreover, Myalgia and self-limiting toxic statin myopathy, rhabdomyolysis, and statin-induced IMNM are 4 clinical phenotypes that SIM can exhibit. 7 The discontinuation of statins in the first 3 cases gives an improvement to the clinical picture, however for statin-induced IMNM, the results may persist or even worsen.2,7 In our case, when atorvastatin was discontinued, there was an obvious improvement in his condition over several months but when rosuvastatin was introduced, the patient experienced a high peak in CK reaching 3393 U/I. For this reason, the persistence of symptoms for months with elevated CK does not immediately exclude the possibility of myotoxicity induced by statins, especially when there is an autoimmune cause.
Serology or a muscle biopsy can provide a definitive diagnosis when taken into consideration with clinical correlation.5,2-7 Serological diagnosis is based on anti-HMGCR antibody testing, which supports the diagnosis of statin-induced IMNM in the appropriate clinical settings with high sensitivity and specificity. 6 However, as mentioned above, antibody positivity is not always found in patients exposed to statins. According to one study, a third of patients with anti-HMGCR-positive IMNM had not been previously treated with statins. 6 Muscle biopsy usually shows necrosis with regeneration of muscle fibers and minor inflammation composed mainly of macrophages.3-4
No international guidelines have yet been published on the treatment of IMNM. Early diagnosis, discontinuation of statins, and in combination with immunosuppressive drugs are considered the cornerstones of treatment. 3 Triple therapy, often consisting of high-dose prednisone (typically 1 mg/kg), intravenous immunoglobulin (IVIG), and an additional drug such as azathioprine or methotrexate have been used successfully in some cases. 1 Some studies have highlighted immunoglobulins as an effective monotherapy, especially in IMNM related to statins. 1
In addition to lifestyle and diet changes, we first tried to reduce the dose of atorvastatin, but the patient’s clinical and biological condition did not improve. Then, we switched to rosuvastatin, which is a hydrophilic statin. According to some research studies, hydrophilic statins are better tolerated than lipophilic statins, but this has not yet been proven by clinical studies. 8 However, the patient continued to experience similar symptoms despite the trial of a hydrophilic statin. In patients like ours who cannot tolerate statins, one can consider proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitor-based treatment such as evolucumab. These medications are well tolerated and useful for primary and secondary prevention of coronary artery disease especially for those who cannot tolerate statin medications. 8
Conclusion
In cases where statin autoimmune-mediated myopathy is considered as a possible differential diagnosis, it is vital to attain a careful history, physical examination, and laboratory work up. In circumstances where the etiology of muscle weakness remains unidentified, one must also consider employing tests such as muscle biopsy, electromyography studies, and autoimmune serology workup. Prompt identification and initiation of treatment is imperative in decreasing morbidity for patients with statin autoimmune-mediated myopathy.
Footnotes
Disclaimer
This work was supported by HCA Healthcare and/or an HCA Healthcare affiliated entity. The views expressed in this publication represent those of the author(s) and do not necessarily represent the official views of HCA Healthcare or any of its affiliated entities.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.