
Abstract
The coexistence of systemic lupus erythematosus (SLE) and myasthenia gravis (MG) is rarely reported, especially the appearance of SLE before MG. In addition to the production of autoantibodies after thymectomy, gene mutation may be an important contributing factor to the overlap of SLE and MG. Here, we report a case of a female patient diagnosed with SLE before MG and found to carry the heterozygous variation COL6A1 c. 2608G>A. We propose that this gene variation weakens the function of COL6A1 and indirectly activates STAT1, resulting in the phenotype of these two autoimmune diseases. This report suggests that it is feasible to explore common pathogenic genes in SLE and MG through future large-scale research.
Introduction
Systemic lupus erythematosus (SLE) is an autoimmune disease linked to genetic factors and estrogen production. 1 Myasthenia gravis (MG) is an antibody-mediated autoimmune disease involving the neuromuscular junction. In MG patients, SLE can appear after thymectomy, 2 while SLE patients rarely develop MG. Hence, a genetic mutation may be involved in the copathogenesis of SLE and MG. Here, we report a case of MG in a parturient patient with SLE and a COL6A1 gene mutation.
Case Report
A 36-year-old Chinese female was diagnosed with SLE after delivery at age 33. At the age of 32, the patient had no symptoms and a blood test revealed a prenatal platelet count of 3 × 109/L. Upon further examination, the nucleolar pattern of anti-nuclear antibody (ANA) was 1:100 (indirect immunofluorescence, IIF). Further testing was negative for routine urine markers, coagulography, complement, anti-neutrophil antibody, anti-cardiolipin antibody, and bone marrow biopsy. Hence, the patient was diagnosed with immune thrombocytopenia with a suspected connective tissue disease. Treatment included intravenous immunoglobulin (20 g ivgtt qd), dexamethasone (10 mg ivgtt qd), and platelet transfusion. After treatment, the platelet number increased to 68 × 109/L. Several days later, the patient delivered a healthy male. Then, 6 weeks later, at the age of 33, the patient presented with face erythema, alopecia, dry eyes and nose, and photosensitivity. Laboratory data revealed the following: ANA, 1:3200 (IIF); C3, 0.76 g/L; C4, 0.09 g/L; anti-SSA antibody, +++; anti-RO52 antibody, +++; anti-dsDNA antibody, 26 IU/ml (ELISA); and anti-nucleosome antibody, positive. Anti-cardiolipin antibody, routine urine markers, serum creatinine, and labial salivary gland biopsy were negative, while leukocyte and lymphocyte counts were decreased with no abnormality in platelet count and bone marrow biopsy. Based on these results, the patient was diagnosed with SLE (SLEDAI-2000
3
score = 9). Treatment included prednisone (1 mg/kg/day), hydroxychloroquine (0.2 g po bid), and intravenous immunoglobulin (20 g ivgtt qd*4 days), which partially relieved all clinical symptoms. Two months after the diagnosis of SLE, there was noticeable improvement in the symptoms of face erythema and alopecia, but ptosis had developed. Laboratory data revealed the following: C3, 0.72 g/L; C4, 0.09 g/L; and anti-dsDNA antibody, 32 IU/ml (ELISA). In addition, leukocyte and platelet counts had decreased, while routine urine markers and serum creatinine were negative. The SLEDAI-2000 score was decreased to 6. Positive neostigmine test, positive AChR antibodies, and single-fiber electromyography confirmed the diagnosis of MG (Osserman classification
4
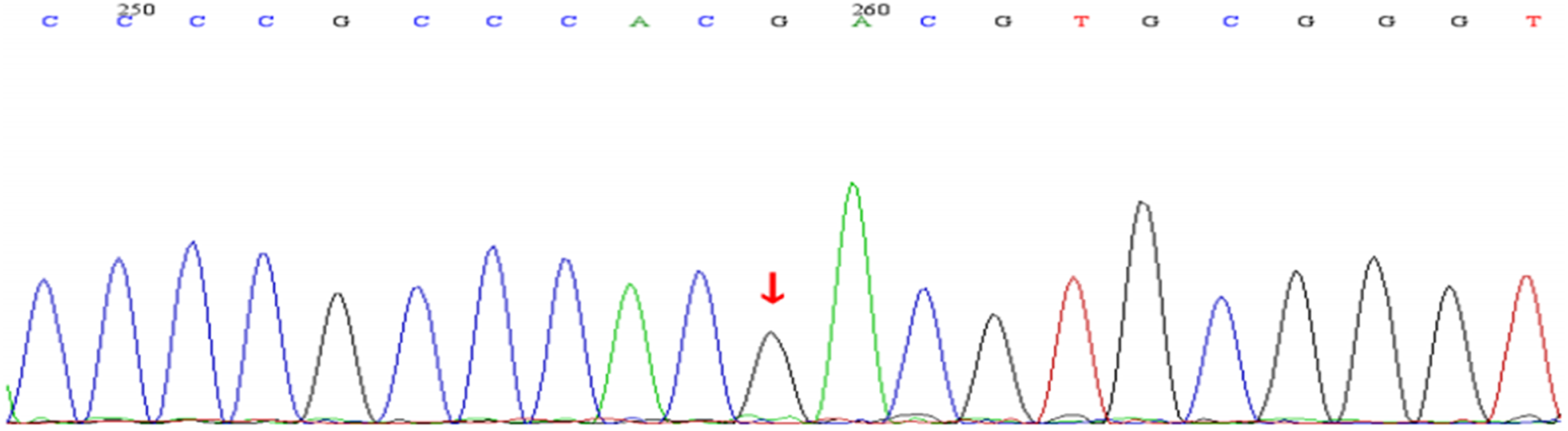
type I). Hydroxychloroquine was discontinued because of possible effects on the neuromuscular junction. Thymectomy was not performed. Treatment was initiated with prednisone (1 mg/kg/day), cyclosporine (50 mg po bid), and pyridostigmine (60 mg po tid). Notably, this strategy was insufficient to control the symptoms of MG but was effective against those of SLE. Six months later, the patient developed bilateral ptosis, dysphagia, and weakness of the extremities. Hence, a potential relationship between SLE and MG was explored. Subsequent whole exome sequencing identified a COL6A1 gene mutation (c.2608 G>A) possibly related to the symptoms of this patient (Figure 1). However, the patient’s mother was healthy and negative for this mutation (Figure 2), while the patient’s father was deceased and no other relatives had similar symptoms. Based on the patient’s symptoms, cyclosporine was discontinued and the following were added to the treatment regimen: plasma exchange; tacrolimus, 2 mg po qd; and the traditional Chinese medicine (TCM) components: Astragalus membranaceus, 60 g; Codonopsis pilosula, 30 g; Atractylodes macrocephala, 15 g; Bupleurum, 15 g; Angelica sinensis, 15 g; Poria cocos, 15 g; tangerine peel, 15 g; Rhizoma dioscoreae, 15 g; and Licorice root, 10 g. This regimen improved most of the patient’s symptoms, although weak vision and limb strength remained. Since then, the following regimen has been maintained: prednisone, 10–20 mg; low-dose pyridostigmin; tacrolimus, 2 mg po qd; and the following TCM components: Astragalus membranaceus, 60 g; Codonopsis pilosula, 30 g; Atractylodes macrocephala, 15 g; Bupleurum, 15 g; Angelica sinensis, 15 g; Poria cocos, 15 g; Tangerine peel, 15 g; Rhizoma dioscoreae, 15 g; and Licorice root, 10 g. At the time of this report, the disorders are in clinical remission. The patient’s gene: red arrow point to the mutational site of COL6A1 c. 2608G>A. The patient’s mother gene: red arrow point is normal.
Discussion
Typically, SLE and MG can coexist or precede one another, although rare associations involving many mechanisms have been proposed in the literature, including the immunostimulatory effect of sex hormones, 5 reduced number of T cells and functional defects, resulting in the generation of autoantibodies, 6 direct effect of anti-malarial drugs on the neuromuscular junction, 7 and the possible involvement of a gene mutation.
This patient developed symptoms at the onset of late pregnancy and was diagnosed with SLE after delivery. Thereafter, she presented with symptoms of MG and was subsequently diagnosed accordingly. In this case, delivery may have triggered SLE due to abrupt changes in sex hormone levels, especially estrogen and prolactin, as sex hormone alterations can influence the differentiation, regulation, and responsiveness of B and T cells, and induce autoimmunity resulting in SLE. 8 Similarly, estrogen-induced production of cytokines and/or immunoglobulins can induce MG postpartum. 9 Thus, changes in female sex hormone levels caused by delivery might be a common trigger of both SLE and MG.
In most MG patients, SLE only appears after thymectomy and a mechanistic relationship between SLE and MG overlap syndrome is suggested by the depletion of T cells. 10 However, this patient developed MG after a diagnosis of SLE and did not undergo thymectomy. We propose that SLE can decrease the number of CD4+/CD25+ regulatory T cells, resulting in functional defects of these cells. Meanwhile, SLE can enhance the activation of Th and B cells, thereby promoting the production of multiple autoantibodies, which can induce the onset of MG. 11 Thus, coexistence of SLE and MG is possibly antibody mediated. Since plasma exchange can effectively remove abnormal antibodies, anti-nuclear and AChR antibodies should be measured in patients of childbearing age with suspected SLE and MG.
Besides, this parturient patient with SLE initially used hydroxychloroquine and 2 months after treatment, developed ptosis. Hence, the physician should be alert to neuromuscular junction symptoms caused by hydroxychloroquine. Given these circumstances, hydroxychloroquine was discontinued in this patient, as there was no improvement in symptoms. Therefore, neostigmine and AChR antibody testing, in addition to electromyography (EMG) were performed to confirm a diagnosis of MG. Notably, MG should be distinguished from the side effects of anti-malarial drugs, and testing of AChR antibodies and EMG should be conducted when MG is suspected in a SLE patient with ptosis.
A recent study found the major histocompatibility complex locus 12 and HLA-DRB1*1602 allele 13 are the primary contributors of most autoimmune disorders, including SLE and MG. However, it remains unclear whether a gene mutation is involved in the coexistence of SLE and MG. The heterozygous variation COL6A1 c. 2608G>A was identified in this patient. The COL6A1 gene is located on chromosome 21q22.3 and encodes the alpha 1 chain of type VI collagen, which is a component of microfibrillar structures 14 . Deletion of the COL6A1 gene leads to a decrease in the volume of tissues containing type VI collagen, such as muscles and tendons. The COL6A1 gene mutation leads to two kinds of genetic diseases: Bethlem myopathy type 1, which is inherited as an autosomal dominant trait with a late age onset and mild symptoms, and Ullrich congenital muscular dystrophy type 1, which is inherited as an autosomal recessive trait with early onset and rapid progression. This patient did not exhibit similar clinical manifestations of the two diseases before the onset of SLE, thus the COL6A1 mutation C. 2608g > A was suspected, as this mutation does not directly cause the occurrence of these two diseases, but rather the susceptibility of SLE patients to autoimmune diseases, such as MG, is due to transmission dysfunction at the neuromuscular junction. The activation of COL6A1 can inhibit STAT1, 14 which is a member of the transcription factor STAT family and involved in cellular responses to cytokines and growth factors, as well as regulation by the immune system. In this patient, the COL6A1 c.2608 G > A mutation gave rise to the amino acid substitution CGA (ARG) > CAA (Gln), which may have weakened the function of COL6A1 and indirectly activated STAT1, resulting in SLE and MG.
Conclusion
We encountered a female patient diagnosed with SLE before MG with the COL6A1 heterozygous variation c. 2608G>A. Future studies are warranted to explore the effect of this mutation on gene function and search common pathogenic genes in SLE and MG through large-scale research. At present, clinicians should consider MG as a potential cause of muscle weakness in women with SLE and investigate causative factors as soon as possible.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was financially supported by Chongqing Science and Technology Commission (cstc2018jxj1130084), Chongqing Postdoctoral Science Foundation (cstc2019jcyj-bshX0044).
Ethics approval
Our institution does not require ethical approval for reporting individual cases or case series.
Informed consent
Written informed consent was obtained from the patient(s) for their anonymized information to be published in this article.