
Abstract
Isoimperatorin (QHS) is a phytoconstituent found in the methanolic extracts obtained from the roots of Angelica dahurica, which contains anti-inflammatory, anti-bacterial, analgesic, anti-tumor, and vasodilatory activities. QHS possesses potent antagonistic activity against lipopolysaccharide (LPS)-induced inflammation; however, the mechanism of action remains unclear. In this study, we investigated the anti-inflammatory effect of QHS and explored the underlying mechanisms. The QHS was purchased from Jiangsu Yongjian Pharmaceutical Co., Ltd. (Jiangsu, China). We performed MTT assay, real-time PCR, ELISA, and western blotting experiments to assess the anti-inflammatory activity and the possible mechanism of QHS in vitro. Molecular docking was performed to study the binding of QHS and myeloid differentiation protein-2 (MD-2) and elucidate the possible anti-inflammatory mechanism. QHS had no significant effect on cell viability. Moreover, pre-treatment with QHS significantly decreased the release of inflammatory cytokines and mediators including NO, TNF-α, IL-6, and IL-1β. In addition, real-time PCR showed that QHS decreased the mRNA expressions of iNOS, COX-2 TNF-α, IL-6, and IL-1β. Western blotting indicated that QHS could inhibit the expression of the proteins associated with the LPS-TLR4/MD-2-NF-κB signaling pathway. Lastly, molecular docking revealed a possible binding mechanism between QHS and MD-2. QHS exhibited anti-inflammatory activity when combined with MD-2, regulating the LPS-TLR4/MD-2-NF-κB signaling pathway, and inhibiting the release and expression of inflammatory cytokines and mediators. Furthermore, QHS can be used as a potential TLR4 antagonist, which blocks MD-2 binding, for treating inflammatory responses induced by LPS.
Keywords
Introduction
Isoimperatorin (QHS), a 6,7-furocoumarin, possesses multiple pharmacological activities, including anti-inflammatory, antibacterial, analgesic, antitumor, and vasodilatory activities (Figure 1). 1 Recent pharmacological studies have shown that QHS and imperatorin inhibit prostaglandin E2 (PGE2) production by suppressing the induction of COX-2 and PGES, thereby exhibiting anti-inflammatory activity. 2 It is reported that imperatorin inhibits LPS-induced NO production through the suppression of iNOS expression in RAW 264.7 cells. Since the induction of COX-2 and iNOS is regulated by similar transcription factors, the mechanism for the suppression of COX-2 expression by imperatorin might be the same as the suppression of iNOS expression. 2 We also learned that QHS could inhibit the production of other inflammatory mediators and inflammatory cytokines, thereby exhibiting anti-inflammatory action.3–5

Chemical structure of QHS.
Lipopolysaccharide (LPS) is a major constituent of the outer membrane of Gram-negative bacteria that stimulates inflammatory reactions,6–8 including the excessive release of inflammatory cytokines and inflammatory mediators.9,10 These mediators can trigger systemic inflammation and cause organ injury. Upon recognition, LPS binding protein (LBP) binds to LPS, and acts on CD14, which is recognized by TLR4-MD-2 through binding in the deep hydrophobic pocket of MD-2 on the cellular surface, leading to the rapid and coordinated activation of various intracellular signaling pathways including activation of NF-κB.11,12 NF-κB, a ubiquitous transcription factor, controls expression of various pro-inflammatory genes. 13 The activation of LPS-TLR4/MD-2-NF-κB signaling pathways triggers systemic inflammation that could cause organ damage and sepsis. MD-2 is the key receptor that activates the LPS-TLR4/MD-2-NF-κB signaling pathway of the LPS-induced inflammatory response. 14 COX-2, an important inducible synthetase in the process of inflammation, is responsible for the production of inflammatory mediators, such as PGE2. 15 COX-2 can be regulated by several upstream inflammatory signaling pathways; however, the TLR4 pathway plays an important role. TLR4 is a key target that mediates inflammation caused by LPS and the inflammatory effect can be exerted by activating and regulating downstream COX-2. NF-κB is the key intermediate link for TLR4 involved in the regulation of COX-2.16,17 Therefore, it is possible that QHS exhibits anti-inflammatory activity by binding to the key target protein MD-2, and regulating the LPS-TLR4/MD-2-NF-κB signaling pathway. However, the mechanism of anti-inflammatory activity of QHS is unclear. In this study, we investigated the mechanism of action of QHS in LPS-TLR4/MD-2-NF-κB signaling the pathway using ELISA, real-time PCR, western blotting, and molecular docking experiments.
The possible anti-inflammatory mechanism of QHS was studied by analyzing the anti-inflammatory activity in vitro and by performing molecular docking experiments. This study was carried out to expound the anti-inflammatory mechanism of QHS and lay a foundation for the research of anti-inflammatory drugs with potential TLR4 antagonists that block its binding to MD-2.
Materials and methods
Chemicals and reagents
QHS (purity ⩾ 98%) was obtained from Jiangsu Yongjian Pharmaceutical Co., Ltd. (Taizhou, Jiangsu, China). Agilent series 1100 instrument was used to check the purity of QHS. RPMI-1640, penicillin-streptomycin, fetal bovine serum, and 0.25% trypsin (1X) were procured from GIBCO (San Angelo, TX, USA). We purchased 3-(4,5)-dimethyl-thiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) from Sigma-Aldrich (StLouis, MO, USA). TRIzol and Fermentas protein marker (10–250 kDa) were obtained from Thermo Fisher Scientific (Waltham, MA, USA). HiScript Reverse Transcriptase (RNase H), 5× HiScript Buffer, 50× ROX Reference Dye 2, SYBR Green Master Mix, and SYBR GREEN Master Mix were purchased from VAZYME (Nanjing, China). DNase/RNase Free water (ddH2O) was procured from GeneCopoeia (Rockville, MD, USA). Ribonuclease inhibitor was obtained from TransGen (Beijing, China). Taq Plus DNA Polymerase, dNTPs, and DL2000 DNA Marker were purchased from TIANGEN (Beijing, China). Random Primer (N6) was acquired from AID-LAB (Xiamen, China). The phosphatase inhibitor, PMSF, RIPA lysate, and BCA Protein Concentration Determination Kit were purchased from Beyotime (Jiangsu, China). N,N,N′,N′-Tetramethylethylenediamine (TEMED) and bromophenol blue were procured from VWR Life Sciences (Radnor, PA, USA). Protein marker (14–120 kDa) was purchased from Beijing Quanjin Biotechnology Co., Ltd. (Beijing, China). Mouse monoclonal antibody β-actin, HRP-labeled goat anti-mouse secondary antibody, and HRP-labeled goat anti-rabbit secondary antibody were purchased from Wuhan Boster Biological Technology Co., Ltd. (Wuhan, China). Rabbit polyclonal antibody TLR4 (96 kDa) and rabbit polyclonal antibody MYD88 (33 kDa) were purchased from Wuhan Sanying Biotechnology Co., Ltd. (Wuhan, China). Rabbit polyclonal antibody P65 (65 kDa) was acquired from Cell Signaling Technology (Danvers, MA, USA). Rabbit polyclonal antibody P-P65 (61 kDa) was obtained from Bioss (Woburn, MA, USA).
Cell culture and treatment
RAW 264.7 cells were obtained from the Type Culture Collection of the Chinese Academy of Science (Shanghai, China). The cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with antibiotics (100 mg/mL penicillin and 100 mg/mL streptomycin) and 10% (v/v) heat-inactivated fetal bovine serum, and incubated in a humidified atmosphere of 5% CO2 at 37°C, 18 until 80% confluency was reached. The medium was changed every 2–3 days.
MTT assay
Cell viability was assessed using an MTT reduction assay. The cells were seeded in a 96-well plate at a density of 5 × 103 cells/well and acclimatized overnight at 37°C. 19 The cells were treated with various concentrations of QHS (8, 40, and 100 μg/mL) for 4 h. The assay is based on the ability of viable cells to reduce blue MTT to formazan, which was measured using a spectrophotometer at 568 nm. The experiment was performed in triplicates.
Cytokine measurement
RAW 264.7 cells were seeded in a 96-well plate at a density of 5 × 103 cells/well and cultured for 24 h in a humidified atmosphere of 5% CO2 at 37°C. The experimental groups were as follows: normal, LPS, and QHS. The QHS group was treated with various concentrations of QHS (8, 40, and 100 μg/mL), followed by 100 ng/mL LPS, and cultured for 24 h. The LPS group was treated with 100 ng/mL LPS. The normal group was left untreated. To determine the cytokine concentration, the cell culture supernatant was analyzed using enzyme-linked immunosorbent assay (ELISA), according to the manufacturer’s protocol.
Real-time polymerase chain reaction (PCR)
Total RNA was extracted from the cells using TRIzol and TURBO DNase. RNA was reverse transcribed using the Superscript first-strand cDNA synthesis kit. 20 The primer sequences used in this experiment are listed in Table 1. The thermal cycling conditions for the PCR reaction were as follows: 50°C for 2 min followed by 40 cycles of 95°C for 10 min, 95°C for 30 s, and 60°C for 30 s. The amplification of PCR products was performed using the primers listed in Table 1. The PCR products were then cloned into the pGEMT vector and sequenced for confirmation. Real-time PCR was performed using the real-time PCR system. 21 The abundance of each gene was determined by comparison with the standard curve. The values of IL-1β, IL-6, TNF-α, iNOS, and COX-2 were normalized to that of β-actin. Each sample was measured three times.
Real-time PCR primer sequences.

Western blotting assay
RAW264.7 cells were lysed using RIPA buffer containing protease inhibitors and phosphatase inhibitors, according to the manufacture’s protocol. The nuclear protein isolation-translocation assay kit was used for separating the nucleus and cytosol proteins, according to the manufacturer’s instructions. Then, the protein concentration was assessed using the BCA Protein Concentration Determination Kit, according the manufacturer’s protocol. 22 Cellular protein extracts were isolated using electrophoresis with 10% SDS-polyacrylamide gel and electroblotted onto PVDF membranes. The membranes were blocked in blocking solution for 2 h at room temperature, followed by incubation overnight with primary antibodies at 4°C. The membranes were washed six times with TBST and incubated with a 1:5000 dilution of HRP-conjugated secondary antibody for 2 h at 37°C. The membranes were washed five times with TBST and developed by treatment with an enhanced chemiluminescence substrate. The band intensities were quantified using Image-Pro Plus (Media Cybernetics, Rockville, MD, USA).
Statistical analysis
Statiatical analysis was performed with the aid of SPSS 17.0 software. The results are presented as the mean ± standard deviation (SD). The statistical significance was analyzed by one-way analysis of variance (ANOVA) followed by Dunnett’s method and Chi-square tests. p < 0.05 was considered significant, and p < 0.01 was considered highly significant.
Docking study
AutoDock Vina (Scripps Research Institute, San Diego, CA, USA) was used to evaluate the binding of QHS with MD-2. 23 The ligand was constructed and performed energy minimization in MMFF94 force field by ChemBioDraw 16.0. The protein structure was prepared in AutoDock 4.2.6 by removal of water molecules and adding hydrogens. The X-ray crystal structure of human MD-2 complexed with lipid IVa (PDB 3vq2, 2.48 Å) was obtained from the Protein Data Bank (pdb:www.rcsb.org). The protonation state of the protein and the ligand were prepared using the default settings. A grid box with dimensions of 60 × 60 × 52 Å (−12.13, 22.813, −2.759) with a spacing of 0.375 Å was constructed around the docking area using the Autogrid 4.2 software. The molecules were docked using Vina with exhaustiveness grade 8. The lowest energy conformations were selected and the ligand interactions with MD-2 were determined. The interaction was visualized using Accelrys Discovery Studio Visualizer 4.0 (Accelrys, San Diego, CA, USA) and PyMOL 0.99.
Results
Cytotoxicity of QHS
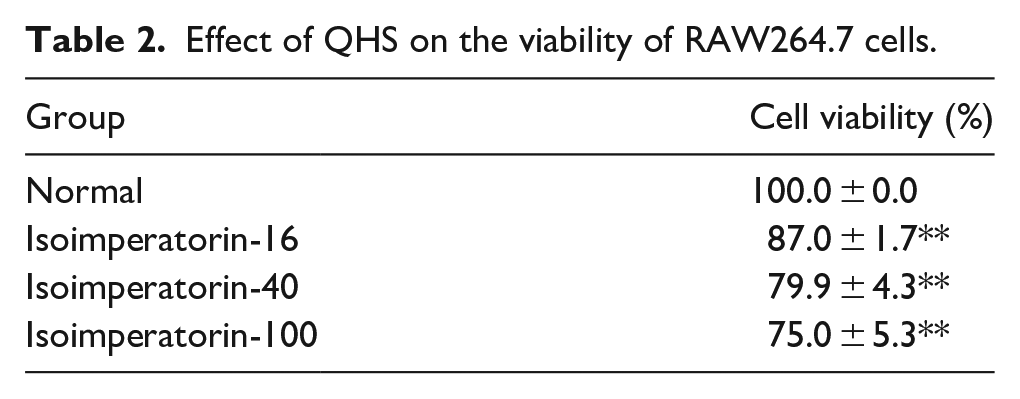
The effect of QHS on the viability of RAW 264.7 cells was determined using MTT assay. QHS showed very low cytotoxicity at the concentrations of 8, 40, and 100 μg/mL compared to that in the control group. Treatment of RAW 264.7 cells with QHS at different concentrations did not significantly affect its cell survival rate (Table 2).
Effect of QHS on the viability of RAW264.7 cells.
QHS inhibits the production of LPS-induced inflammatory cytokines and mediators in RAW 264.7 cells
NO, TNF-α, IL-6, and IL-1β are the key inflammatory molecules that are released in response to LPS. As shown in Figure 2, the levels of inflammatory molecules were significantly higher in the LPS group than those in the control group (p < 0.01). Moreover, compared to the LPS-induced control group, the QHS treatment significantly attenuated the release of inflammatory cytokines and mediators (p < 0.05 and p < 0.01) (Figure 2).

Effect of QHS on the inflammatory molecules produced by RAW 264.7 cells. The levels of NO, IL-1β, IL-6, and TNF-α in the cell culture supernatant were measured using ELISA.
QHS inhibits LPS-induced mRNA expression of iNOS, TNF-α, IL-6, IL-1β, and COX-2 in RAW 264.7 cells
The experiments demonstrated that 100 μg/mL QHS showed anti-inflammatory activity by decreasing the production of TNF-α, IL-6, and IL-1β in LPS-activated RAW264.7 cells. Moreover, iNOS and COX-2 are important targets for the LPS-induced inflammatory response. Therefore, we performed to investigate the mRNA expression of iNOS, TNF-α, IL-6, IL-1β, and COX-2 in RAW 264.7 cells. The groups were the same as that the previous experiment. Total RNA was extracted from the cells after treatment with TRIzol and DNase and the mRNA expressions of TNF-α, IL-6, IL-1β, iNOS, and COX-2 in RAW 264.7 cells were evaluated. We found that the mRNA expression levels of TNF-α, IL-6, IL-1β, iNOS, and COX-2 in the LPS group were significantly higher than those in the normal group (p < 0.01), while the mRNA expression levels in the QHS group were significantly lower than those in the LPS group (p < 0.01) (Figure 3).

Effect of QHS on cytokine mRNA expression in RAW 264.7 cells. The expressions of iNOS, IL-1β, IL-6, TNF-α, and COX-2 were determined.
QHS inhibits LPS-induced expression of related proteins in the LPS-TLR4-NF-κB signaling pathway in RAW 264.7 cells
TLR4 is responsible for LPS-induced signal transduction leading to NF-κB activation. NF-κB is a ubiquitous transcription factor that governs the expression of several proinflammatory and immune system-modulating genes. The LPS-TLR4/MD-2-NF-κB signaling pathway plays an important role in the inflammatory response induced by LPS. To confirm the anti-LPS mechanism of QHS in the regulation of the LPS-TLR4/MD-2-NF-κB signaling pathway, we measured the protein expression levels of TLR4, MyD88, NF-κB p65, NF-κB p-p65, and iNOS in RAW 264.7 cells. The levels of TLR4, MyD88, NF-κB p65, NF-κB p-p65, and iNOS were upregulated in the LPS group (p < 0.01) compared to those in the normal group. On the contrary, the levels were significantly reduced in the QHS group (p < 0.01) compared to those in the LPS group (Figure 4(b)).
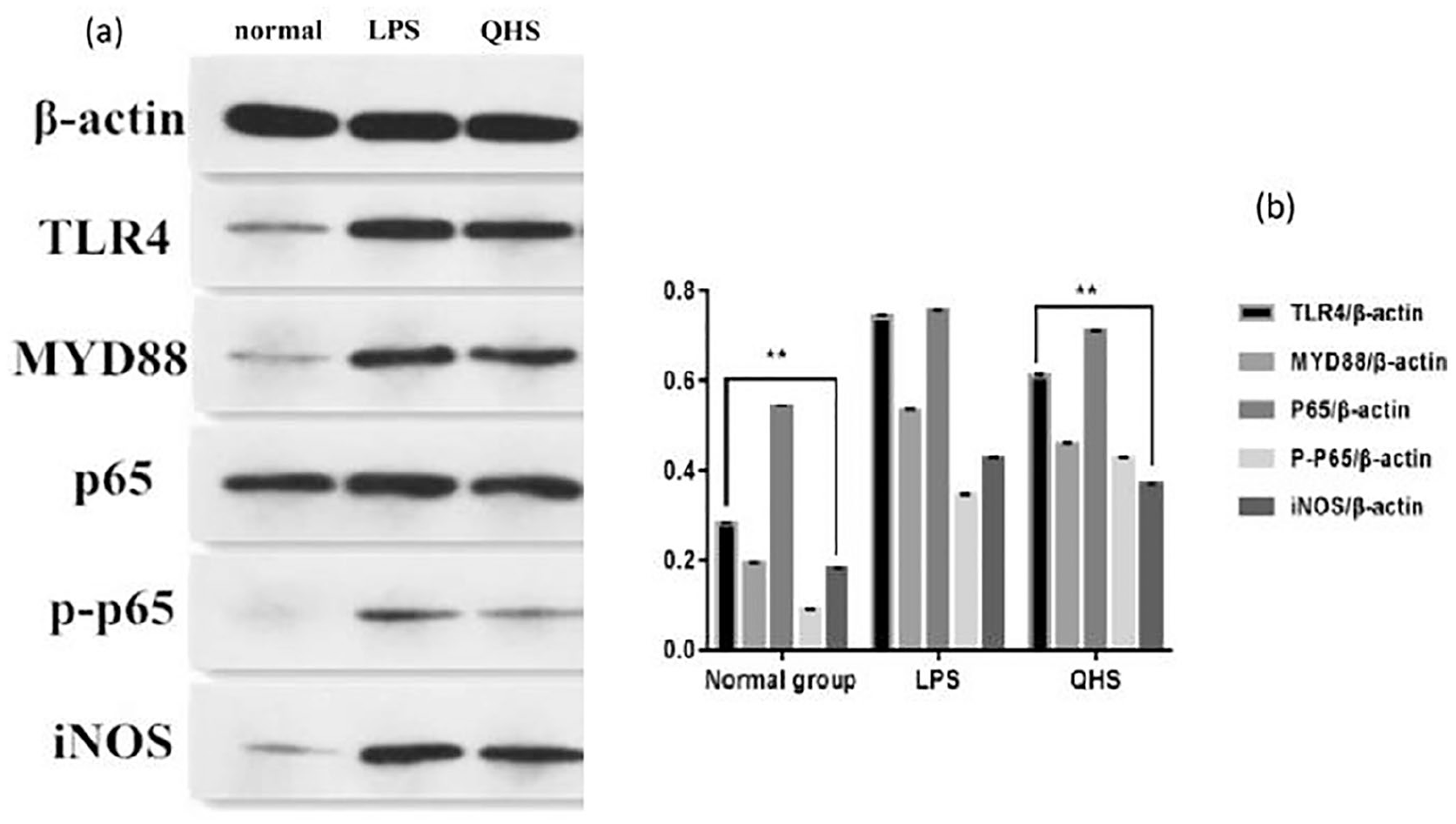
(a) Western blotting analysis of TLR4, MyD88, NF-κB p65, NF-κB p-p65, iNOS, and β-actin protein expression and (b) Effect of QHS on the expression of related proteins in the signaling pathway. TLR4, MyD88, NF-κB p65, NF-κB p-p65, and β-actin protein expressions were determined using western blotting. Band intensities were quantified from three independent experiments.
Molecular docking of QHS
To explore the interaction between QHS and MD-2, the QHS molecule was docked in the crystal structure of MD-2 using AutoDock with a binding affinity of −6.7 kcal/mol. Two phenolic ether groups and one carbonyl group in QHS formed hydrogen bonds with Ile-120, Val-63, and Tyr-102 in the MD-2 binding site, respectively. The whole QHS molecule, which was surrounded by hydrophobic groups, was bound inside the hydrophobic pocket of MD-2 (Figure 5). The core of QHS, 7H-furo[3,2-g]chromen-7-one, was inserted into the MD-2 pocket and interacted with interior hydrophobic residues, such as ILE52, ILE54, PHE121, and PHE126. The isobutenyl group occupied the hydrophobic site formed by GLU437 and ARG90. These interactions suggested a possible binding mechanism between QHS and MD-2.

Molecular modeling of QHS bound to MD-2 obtained in the docking study.
Discussion
The development of structural crystallography and computational methods has facilitated the identification of three-dimensional structures of several disease-related proteins. Recently, structure-based drug screening has been gaining increased attention and when compared with traditional drug discovery, structure-based drug screening could reduce the experimental cost and accelerate the process of drug development. We developed anti-inflammatory drugs based on MD-2 protein and found that QHS showed efficient binding with MD-2. Therefore, we performed in vitro experiments to evaluate the anti-inflammatory activity of QHS along with molecular docking to investigate the possible mechanisms involved in binding of QHS and MD-2. MTT assay revealed that different concentrations of QHS had no significant effect on cell viability. Thus, we studied the anti-inflammatory activity of QHS to investigate the release and expression of inflammatory cytokines and mediators. LPS specifically binds to the corresponding receptor proteins on the surface of macrophages, thereby activating the LPS-TLR4/MD-2-NF-κB signaling pathway and promoting the release of NO, TNF-α, IL-6, and IL-1β to participate in the inflammatory reaction.24,25 Nitric oxide synthase is an essential enzyme for NO synthesis and is divided into primary nitric oxide synthase and iNOS. 26 iNOS is mainly produced under pathological conditions and induced by proinflammatory substances and inflammatory mediators. The expression of iNOS could be increased, thereby inducing the production of large amounts of NO. Therefore, inhibition of the release of NO or expression of iNOS may be an important target for the treatment of inflammatory diseases. LPS-induced macrophage cell inflammation models are widely used to study inflammatory responses. We evaluated the release of NO, TNF-α, IL-6, and IL-1β to determine the anti-inflammatory activity of QHS. When compared with the LPS-induced control group, 100 μg/mL QHS significantly inhibited the release of TNF-α, IL-6, IL-1β, and NO in LPS-induced RAW264.7 cells. This result indicated that QHS inhibits the production of inflammatory cytokines, such as TNF-α, IL-6, and IL-1β, and mediators, such as NO, induced by LPS and reduces the inflammatory response.
COX is the key rate-limiting enzyme for the synthesis of various prostaglandins and contains two isozymes, COX-1 and COX-2. COX-1 is present in normal tissues and plays an important physiological role in protecting the gastrointestinal mucosal cells and maintaining the normal function of platelets and the kidneys. However, COX-2 expression is generally lower in normal tissues and its expression is mainly associated with inflammation in cells or tissues. Inflammatory cytokines stimulate COX-2, which further exacerbates the inflammatory response. The expression level of COX-2 is related to the severity of inflammation.27,28 COX-2 gene-deficient rats resisted infection and death mediated by LPS and showed delayed leukocyte infiltration into organs. 29 We used real-time PCR to detect the mRNA expression levels of TNF-α, IL-6, IL-1β, iNOS, and COX-2. The results showed that QHS could significantly reduce the mRNA expression of TNF-α, IL-6, IL-1β, iNOS and COX-2, thus decreasing the inflammatory response.
The transcription factor NF-κB is mainly located in the plasma and belongs to the IκB protein family. 30 Activation of the NF-κB/IκBα complex depends on the phosphorylation of two conserved serine residues, which are located at the N-terminus of the IκB protein. The phosphorylation of the residues results in polyubiquitination of the IκB protein, which leads to the degradation of the IκB protein mediated by the 26S proteasome. 31 The protein degradation activates the NF-κB signaling pathway, leading to the accumulation of NF-κB in the nucleus.32–34 NF-κB can also be activated by pro-inflammatory factors, bacteria, LPS, and viruses. The activated NF-κB binds to the specific DNA sequence of the target gene, which mediates the transcription of more than 400 genes associated with inflammation, immune regulation, tumor cell proliferation, invasion, metastasis, angiogenesis, and radiochemotherapy tolerance.35–39 After TLR4 activation, LPS activates NF-κB via the MyD88-dependent pathway, which promotes the release of downstream TNF-α, IL-6, NO, and other inflammatory cytokines, and triggers the inflammatory response. We used western blotting to investigate the related proteins in the LPS-TLR4/MD-2-NF-κB signaling pathway of QHS. The results showed that QHS inhibited the protein expressions of TLR4, Myd88, P65, P-P65, and iNOS in the LPS-TLR4/MD-2-NF-κB signaling pathway.
The recognition of LPS by monocytes and macrophages and their signal transduction are essential as a defense response to Gram-negative infections in the host. LBP interacts with LPS, and acts on the CD14 membrane receptor of monocytes and macrophages. This signal is then transduced into the cell by the corresponding signaling molecules,40–42 causing activation or damage to the cell. However, the CD14 molecule is a glycoprotein anchored to the surface of the membrane, and because it lacks transmembrane and intracellular regions, it cannot transmit signals into cells by itself. Therefore, the synergistic action of toll-like receptor 4 (TLR4) and myeloid differentiation protein-2 (MD-2) is required for intracellular transmission of the stimulation signal induced by the bacteria. 43 The discovery of TLR4 and MD-2 proteins was a major breakthrough in the understanding of LPS receptor recognition mechanisms. However, further studies have found that in the presence of only TLR4, without MD-2, the cells do not respond or respond weakly to LPS. This indicates that TLR4 must be assisted by MD-2 to recognize LPS and mediate further inflammatory responses. Blocking the binding of LPS to MD-2 is nearly 100 times easier than blocking the binding of LPS to TLR4/MD-2.9,44 Therefore, a compound that can interfere with the binding of LPS to MD-2 or inhibit the binding of MD-2 to TLR4 might be a more efficient and cost-effective candidate for controlling the LPS-induced inflammatory response. Therefore, in this study, we used MD-2 as the target and QHS to find TLR4 antagonists and examined the binding of MD-2 and QHS using molecular docking. The result exhibited that QHS regulated LPS-TLR4/MD-2-NF-κB signaling pathway by binding to MD-2, inhibited the release and expression of cell cytokines and mediators, and decreased the expression of the proteins related to LPS-TLR4/MD-2-NF-κB signaling pathway. However, there are a few drawbacks in this study, for instance, the in vivo experiments are further required to elucidate the anti-inflammatory mechanism of QHS.
Conclusion
In summary, QHS exhibits anti-inflammatory activity by blocking the key protein MD-2 in the LPS-TLR4/MD-2-NF-κB signaling pathway and inhibits the release and expression of inflammatory cytokines and mediators. This study laid a foundation for further studies on the anti-inflammatory effects of QHS.
Footnotes
Acknowledgements
The authors thank the reviewers for their useful comments and the assistance provided by the SAGE language Services.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The study was supported by funding obtained from the Natural Science Foundation of China (No. 81303205), Liaoning University Innovation Talent Support Program (LR2017002), and Shenyang Science and Technology Plan Project (18-013-78).