
Abstract
Inflammatory polyradiculoneuropathies are heterogeneous disorders characterized by immune-mediated leukocyte infiltration of peripheral nerves and nerve roots leading to demyelination or axonal degeneration or both. Inflammatory polyradiculoneuropathies can be divided into acute and chronic: Guillain–Barré syndrome and chronic inflammatory demyelinating polyneuropathy and their variants. Despite major advances in immunology and molecular biology have been made in the last years, the pathogenesis of these disorders is not completely understood. This review summarizes the current literature of the clinical features and pathogenic mechanisms of inflammatory polyradiculoneuropathies and focuses on current therapies and new potential treatment for the future.
Keywords
Introduction
Inflammatory polyradiculoneuropathies are immune-mediated disorders that affect the peripheral nerves and nerve roots, thus resulting in demyelination or axonal degeneration and sensory-motor deficits.1,2 Inflammatory polyradiculoneuropathies can be divided into two major subgroups (acute and chronic): Guillain–Barré syndrome (GBS), chronic inflammatory demyelinating polyneuropathy (CIDP), and their variants.3,4
GBS is an immune-mediated disorder characterized by rapid progression of inflammatory demyelination and axonal injury to the nerve roots and peripheral nerves. In the last years, the role of humoral immunity has grown. 3 The most shared theory describes antibodies arising from molecular mimicry between the lipooligosaccharides of microorganisms and surface molecules of the peripheral nerves.2,3
CIDP is believed to have autoimmune pathogenesis in which both cellular and humoral immunity work together damaging the peripheral nerve, against antigens in the Schwann cell or myelin and antigenic targets at the node of Ranvier (NOR). 4
Overall, the immunopathogenesis and definitive mechanisms of inflammatory polyradiculoneuropathies have yet to be well-characterized and current knowledge consists mainly of hypotheses derived from observational human in situ, human in vitro and animal in situ/in vivo studies. Many antibodies against proteins have been recently identified in the NOR, which are short gaps in the myelin sheath, where a rapid propagation of the action potentials occurs.5,6
Also, recent evidence suggests that molecules in the compact myelin, but also within the nodal or paranodal regions may be probable targets of the immune attack in CIDP.5,6 Furthermore, individual susceptibility, genetic and environmental factors are emerging as a possible determinant in inflammatory neuropathies.1,2 It is necessary to better understand peripheral neural-immune interactions and support the development of selective drugs (i.e. drugs active on specific leukocyte subpopulations) that may offer better options to achieve improved outcomes.2,7
In this review, we discuss the best evidence on GBS and CIDP pathogenesis, and we report new perspectives focusing on possible therapeutic applications.
Guillain–Barré syndrome
Definition
Guillain–Barré syndrome is an acute immuno-mediated disorder characterized by rapidly progressive and self-limiting sensory-motor deficits (within 4 weeks) due to inflammatory demyelination or axonal injury (or both) to the nerve roots and peripheral nerves. The incidence is between 1 and 2 cases for 100,000 people slightly more prevalence in males.3,8 In approximately 70% of the cases, there is a history of upper or lower respiratory illness or gastroenteritis in the preceding 10–14 days. 3 A wide range of microorganisms, including Cytomegalovirus, Mycoplasma pneumoniae, Epstein–Barr virus, Influenzae A, Haemophilus influenzae, Enterovirus, and Campylobacter jejuni have been associated with GBS, showing a seasonal trend, that is, in autumn for Campylobacter jejuni and in the spring for winter Influenza virus. 9
Clinical symptoms and subtypes
Typical GBS presents as rapidly progressive bilateral ascending areflexic paralysis. Additional symptoms can occur ranging from mild sensory involvement, cranial nerve paralysis (i.e. bilateral facial weakness) to autonomic dysfunction and radicular low back pain.
Since its early description, several variants of GBS have emerged including acute inflammatory demyelinating polyradiculoneuropathy (AIDP), the most frequent type, acute motor axonal neuropathy (AMAN), acute motor-sensory axonal neuropathy (AMSAN), Miller Fisher syndrome (MFS), and the pharyngeal-cervical-brachial presentation. Finally, acute autonomic and acute sensory neuropathies are rare and poorly defined forms of GBS. 10
In AIDP sensory symptoms are moderate with the presence of pain, autonomic dysfunction, and signs of cranial nerve dysfunction. Nerve conduction studies show demyelinating features. 11 In AMAN, on the contrary, the motor fibers injury is more prominent, the sensory fibers are preserved; cranial nerves paresis and pain are less frequent, but there are a more rapid progression and a slow recovery. 12 Electrophysiology in AMAN shows an axonal neuropathy. 11 In AMSAN sensory involvement is added to the clinical picture of AMAN.
MFS is characterized by the typical triad of ophthalmoplegia, ataxia and areflexia and is featured by the presence of anti-GQ1b antibodies.13,14 The last form, less frequent, is the pharyngeal–cervical–brachial presentation, a localized form of oropharyngeal and cervicobrachial weakness associated with areflexia.3,15
Finally, there are neuropathies with an acute onset mimicking GBS cases with profound sensory and/or autonomic impairment in the absence of any infectious or toxic etiology; these conditions are believed to follow immune mechanisms similar to those in GBS causing a neuronopathy in the sensory and/or autonomic ganglia.16,17
Pathogenesis
GBS is a complex and heterogeneous disorder, which follows multiple triggering events in the majority of cases, ranging from infections, vaccines, and surgery. The pathogenesis differs for each subtype of the disease. Although both branches of the immune system (T and B cells) play a role, the role of humoral immunity has grown in the recent past and now GBS is considered an antibody-mediated disorder in the AMAN and MFS subtypes.1,2,8 Specific antibodies against gangliosides (GM1, GD1a, and GQ1b) were reported in the AMAN and MFS. 8 In AMAN, IgG antibodies against membrane gangliosides GM1 and GD1a have been identified, and they are proven to attack axons of motor fibers ate the NOR, thus explaining prominent axonal features in nerve conduction studies. However, not all antibodies binding gangliosides at the NOR activate myelin-destroying complement. 18
Nerve injury appears to be the result of molecular mimicry between microbial and nerve antigens. The glycans expressed on lipooligosaccharides of infectious agents are supposed to trigger a humoral response against similar carbohydrate antigens in the peripheral nerves. Interactions between the immune system and microorganisms are not still understood, but individual susceptibility and environmental factors could interact with each other altering immune-tolerance.3,8 Until now, only molecular mimicry between C. jejuni lipooligosaccharide and GM1 and GD1a has been found.19,20
Despite its higher prevalence, AIDP remains less understood than more rare variants of GBS. Many infectious agents have been linked to demyelinating forms of GBS and common target antigens have not been found yet. Anyway, the presence of antibodies in AIDP may be argued from complement activation, myelin destruction and phagocytosis by macrophages.
The occurrence of GBS in rare family cohorts suggests a role of genetic factors in disease susceptibility. 1 However, many genes implicated in the pathogenesis of GBS have been investigated with negative or conflicting results. 21
It seems that IgG1 and IgG3 subclass immunoglobulins could bind gangliosides GM1 and GD1a. They can allow complement fixation, thus attracting macrophages with an increase in phagocytic activity and depositing membrane attack complexes on the axolemma. 22 This mechanism may lead to reversible conduction block or, in the most serious cases, can cause severe axonal degeneration with poor recovery.5,11
Antibodies against proteins in the NOR, including gliomedin, contactin, TAG-1, moesin, and neurofascin have been identified. 22 In particular, antibodies against moesin have been found in AIDP during cytomegaloviru (CMV) infection. In very few cases, antibodies against the glycolipid LM1, sulphoglucuronosyl paragloboside, galactocerebroside, and sulfatide have been found. 23
However, a wide range of infectious agents can induce the production of antibodies in AIDP, it was difficult to find a common antigenic stimulus and to identify specific antibodies for the disease.
GBS may also occur during a primary HIV infection, at the time of seroconversion or within the first 3–6 months after the start of antiretroviral therapy, generally in the presence of high CD4+ T-cell counts.
It may be the result of a direct action of HIV-1 virus on the nerves, or autoimmune mechanisms, with the formation of antibodies against myelin. However, the outcome of GBS in these patients is often favorable. 24
Diagnosis
The diagnosis of GBS is mainly clinical: progressive weakness in the four limbs with hypo/areflexia is the most common presentation. Additional symptoms and signs occur according to the different clinical subtypes.
Further investigations may be useful or even necessary for the diagnostic confirmation of GBS. 25 Cerebrospinal fluid (CSF) is very important to exclude other causes of symmetric weakness, usually associated with an increase in CSF cell count. 25 In fact, GBS is typically associated with albumin-cytologic dissociation, the combination of a normal CSF cell count with increased protein level.
However, a normal CSF protein concentration does not exclude GBS, especially in the first week after the onset of the disease.
High titers of anti-GM1, anti-GD1b, anti-GD1a, and anti-GalNAc-GD1a ganglioside antibodies of the IgG class are frequently reported in GBS. However, specific serological tests have a limited role in the diagnosis of GBS, except for MFS, in which elevated anti-GQ1b ganglioside antibodies are consistently found in about 95%–98% of patients. 14
Electrophysiological studies can show demyelinating features with motor conduction blocks in peripheral nerves in demyelinating subtypes of GBS. Earlier in the course of the disease, electrophysiology could be normal or only mild altered. We can find only prolonged latency or absent F waves (Figure 1). In axonal subtypes of GBS, there may be a decrease in Sensory Action Potentials (SAPs, in AMSAN) and/or compound motor action potentials (CMAPs) amplitudes (in AMAN and AMSAN); in AIDP, increased distal motor latencies, slowed motor conduction velocity, temporal dispersion and partial or total motor conduction blocks are observed.11,25,26
Nerve conduction studies in Guillain–Barré syndrome. Electrodiagnostic studies performed 3 weeks from onset in a GBS patient; in (a) prolonged distal motor latency, temporal dispersion with polyphasic CMAP of the peroneal nerve; in (b) rare F-waves with prolonged latency.
Nerve conduction studies, even if useful in the differential diagnosis among GBS subtypes, are not mandatory to obtain a GBS diagnosis. Clinical and instrumental definition and guidelines for the diagnosis of GBS were elaborated by the Brighton Collaboration. 27
Outcome
In GBS, the course is usually monophasic and relapses are rare. The prognosis for most patients with GBS is favorable with most of the improvement in the first year. Approximately 87% fully recover or report minor sequelae, especially weakness, numbness, and persistent fatigue. 28 A minority of patients have relevant disability and pain. Advanced age of onset, previous diarrhea or C. jejuni infection, the early need for intubation and ventilator support during the illness, and severe weakness are considered negative prognostic factors. Mortality in GBS range from 3% to 7% and is due to respiratory failure, infections, deep vein thrombosis or autonomic dysfunction (i.e. wide blood pressure swings and cardiac arrhythmias).3,25,28
Treatment
GBS is a potentially life-threatening disease. Patients require close monitoring to prevent and treat any complications, such as dysphagia, deep vein thrombosis, dysautonomia, respiratory failure, and pain.
Intravenous immunoglobulin (IVIg) and plasma exchange (PE) are equally effective as first-line treatments. 29
IVIg is a formulation obtained from plasma pooled from thousands of donors, containing antibodies (mostly IgG, subclass distribution identical to that of normal human serum but small percentages of IgA are also present) directed both against foreign pathogens and self-antigens. Other components of IVIg preparations are sugars (such as sorbitol, glucose, and sucrose), sodium, amino acids, and low titers of isohemagglutinin antibodies (anti-A, anti-B, anti-C and anti-E blood group). 30
Despite their large use in clinical practice, the proposed mechanism of action of IVIg is only partially known. Suppression of the inflammatory and immune-mediated responses trough neutralization of antibodies, fragment crystallizable (FC) receptor blockade, or B- or T-cells immune-regulation are the presumed mechanisms involved. 8
IVIg side effects are mild and infrequent (less than 10% of patients) and include mild to moderate headache, fever, chills, and myalgias. Chest or back pain during the first few hours, usually responding to stopping and slowing the infusion, is also common, as well as post-infusion fatigue, fever, and/or nausea. Thromboembolic events (i.e. strokes, pulmonary embolism, or myocardial infarction) are rarely observed and are caused by an increase in serum viscosity. Patients with IgA deficiency may experience a severe anaphylactic reaction due to the presence of low levels of IgA in common IVIg preparations and anti-IgA antibodies in serum of such patients. Other rare reactions are acute renal tubular necrosis, skin reactions (i.e. urticaria, lichenoid cutaneous lesions, pruritus of the palms, petechiae, and eczema), and severe headache. 31
A standard IVIg regimen of 2 g/kg bodyweight (0.4 g/kg/d for 5 days) is recommended in patients unable to walk unaided, within the first 2 weeks after onset of symptoms. 3 It remains unknown whether administrating IVIg over 2 days (1 g/kg per day) is equivalent or superior to the standard 5 days (0.4 g/kg per day) protocol.
However, early relapses have been shown to occur in patients with an initially good response. This fluctuating course is considered to be induced by the temporary effect of IVIg or PE on the disease process. The expression “treatment-related fluctuations” (TRFs) was coined to avoid confusion with the relapsing course of CIDP.32,33 TRFs occur more often in shorter IVIg regimen. 34
Compared to PE, IVIg are easier to administer, have fewer side-effects and are better tolerated in children. 35
PE removes antibodies and other possible inflammatory components, such as complement, and may improve T-cell suppressor function.8,36 PE consists of five sessions (total amount of fluid exchanged of 200–250 mL/kg, 40–50 mL/kg per session) over 2 weeks. As PE is the most effective when started within 7 days of symptom onset, it should start as soon as possible from onset.8,36 Hypotension, arrhythmias and bleedings are the principal PE side effects. According to the Cochrane review, PE increases the chance of complete muscle strength recovery after 1 year when compared to supportive care alone, although there is probably a slight increase in the risk of relapse. 36
About 10% of patients treated with either PE or IVIg show no clinical response or deteriorate after initial improvement. TRFs are usually managed with a second course of IVIg or PE with benefit. 32 However, three or more episodes of deterioration after 8 weeks from onset, may suggest the diagnosis of acute CIDP. In this case, the therapeutic approach should be reconsidered, usually needing chronic maintenance treatment with IVIg or a switch to corticosteroid treatment. 37
Whether GBS patients with a poor prognosis may benefit from a second course when administered 1-week after the start of the first course of IVIg is the principal aim of the ongoing GBS- SID trial, whose first results are expected soon. 38
There is no evidence that the combined use of PE before IVIg, may improve the outcome. 39 On the contrary, PE after IVIg should be avoided because of the possible wash out of IVIg. 40
Both oral and intravenous corticosteroids, alone or in combination with IVIg or PE, have not shown benefit in long terms. 41
Chronic inflammatory demyelinating polyneuropathy
Definition
CIDP is a rare disease with a range of prevalence between 1.97 and 4.77 per 100,000.4,42,43 It is characterized by progressive or relapsing motor or/and sensory symptoms and reduced or absent muscle stretch reflexes. 44 The onset is typically insidious, usually evolving over 8 weeks with a relapsing course, or acute with a chronic progressive or relapsing course. In a minority of patients, a “GBS-like” presentation have been reported. Prominent sensory symptoms, as well as the absence of autonomic dysfunction are typical of CIDP, while facial weakness, cranial nerve deficits, and respiratory failure suggest GBS. 37
CIDP variants: the atypical CIDP
CIDP has a wide spectrum with heterogeneity of clinical phenotypes. Typical CIDP is characterized by progressive or relapsing motor and sensory dysfunction of the peripheral nerves, usually involving the four limbs with hypo- or areflexia, developing over at least 2 months. However, only 50% of CIDP patients present a typical form, so that “atypical CIDP” has been distinguished for different patterns of motor and sensory involvement: asymmetrical, multifocal, and only distal limb involvement.4,45,46 The clinical importance of atypical CIDP identification derives from the fact that they usually have a worse response to therapy. According to case series and multicentre studies, the more frequent atypical variant is the distal acquired demyelinating sensory (DADS) neuropathy.45,47 The multifocal acquired demyelinating sensory and motor neuropathy (MADSAM or Lewis–Sumner syndrome) and the pure sensory and motor CIDP are the last types. 48 DADS has predominantly distal sensory symptoms, and conduction studies show distally accentuated demyelination findings. MADSAM presents asymmetric motor and sensory involvement with electrophysiologic evidence of multifocal demyelination. Motor and sensory CIDP are characterized by the absence or reduction of deep tendon reflexes in all limbs associated with motor or sensory fibers demyelination, respectively.45 –47
Pathogenesis
Current knowledge supports autoimmune pathogenesis of CIDP in which both cellular and humoral immunity, against Schwann cell or myelin antigens, damage the peripheral nerve.4,44
Animal models with a spontaneous autoimmune polyneuropathy (SAP) that mimics the progressive form of CIDP have been developed in the last decade. These experimental models showed the relevant role of regulatory T and B lymphocytes (Tregs, Bregs).49,50 However, the immunopathogenesis and definitive mechanisms have yet to be well characterized.
Cellular-mediated demyelination is supported by invasion and loss of myelin lamellar structure by T-cell and macrophages. 44 On the other side, there is little evidence of humoral immunity in CIDP. Several CIDP nerve biopsies show immunoglobulin and complement fixation.4,44 Besides, the early improvement after PE raises the possibility of an antibody-mediated ion channel alteration at the NOR, consistent with a rapid reversal, rather than a myelin or axon repair process, requiring longer time. 51 These findings support a role of antibodies, but their specific target is lacking.
Disease susceptibility has been studied in CIDP patients through a wide analysis of possible genes implicated in the pathogenesis. In a study on human leukocyte antigen (HLA) associations in patients with CIDP, the gene frequency, and the frequency of individuals positive for HLA-DR2 were greater in female patients than female controls. 21 Another association has been found with a homozygous genotype for a low repeat number of tandem GA in the SH2D2A gene, implicated in the control of early T-cell activation. This genotype could result in defective control and elimination of autoreactive T-cells. 52 More recently, DRB1*15 alleles have been related to anti-NF155 + CIDP patients. 53
Cellular factors
During active disease, CD4+ T-cells in the periphery upregulate activation markers such as t-bet and pstat175 and secrete pro-inflammatory cytokines including interleukin (IL)-2, interferon γ (IFNγ), IL-1, the chemokines interferon gamma-induced protein (IP) and macrophage inflammatory protein 3 β (MIP3β).
This release of cytokines and chemokines into the circulation causes further activation of macrophages and induces upregulation of the vascular cell adhesion molecule (VCAM)-1, endothelial leukocyte adhesion molecule (ELAM) and intercellular adhesion molecule (ICAM) on endothelial cells lining the blood vessels of the nerve. 4
The macrophages and the Schwann cells, expressing the co-stimulatory molecules B7-1and B7-2, play an important role in local antigen presentation to CD4+ T-cells, that express their counter-receptors of cytotoxic T-lymphocyte associated protein 4 (CTLA-4) and CD28. 54
Activated T-cells adhere to the endothelial cells, roll along the vessel surface and then migrate across the blood-nerve barrier (BNB). Prolonged secretion of inflammatory mediators may increase BNB permeability, thus allowing soluble factors (such as antibodies) to access the endoneurium. Breakdown of the BNB can be visualized by magnetic resonance imaging (MRI) gadolinium enhancement of nerve trunks or plexuses in patients with CIDP.55,56
The presence of an antigen-driven T-cell response against peripheral nerve antigens in CIDP is confirmed by studies showing that the few CD8+ and CD4+ T-cells found in the nerve biopsies have the same oligoclonal restrictions of the peripheral blood lymphocytes. 57
Furthermore, macrophages play an active role in many aspects of the immune response such as antigen presentation, the release of pro-inflammatory cytokines and represent the final effector cells associated with demyelination. They penetrate the basement membrane of the Schwann cell, displace the cytoplasm, split the myelin lamellae with focal destruction of the myelin sheath causing the so-called “macrophage-mediated demyelination.” 54
The role of cytotoxic T-cells, on the other hand, is controversial. In CIDP nerves, Schwann cells up-regulate MHC class I molecules, potentially enabling the reactivation of cytotoxic T-cells (CD8+). These CD8+ T-cell clones are enriched in the nerve suggesting that an antigen-driven CD8+ cell-mediated attack on the nerve contributes to the pathogenesis of CIDP.
However, evidence of these CD8+ T-cells in direct contact with their target cells in situ is lacking, limiting further conclusions about their role as cytotoxic effector cells. 4
In CIDP, to date, no foreign or self-antigen has been yet identified as a CD8+ target, but there is evidence of the similar clonal expansion of CD8+ cells both in sural nerve and peripheral blood. 57
Recent evidence from spontaneous animal models has demonstrated the role of Tregs and Bregs in the pathogenesis of CIDP.49,50 Adoptive transfer studies revealed that Tregs suppressed SAP, while Bregs attenuated disease severity. Moreover, B-cell deficiency in the mice model prevented the development of SAP, which would indicate that the pathogenic role of B cells predominates over its regulatory role in this model. 49
Humoral factors
In CIDP humoral factors may play an important role owing to the beneficial effect of PE. 58 Autoantibodies are found in approximately 30% of patients with CIDP, although specific antigens have been identified in no more than 13% of cases. 23
The presence of complement-fixing IgG and IgM deposits on the myelin sheath 59 provided the first evidence of the antibody’s involvement. 60 Sural nerve biopsy specimens from patients with chronic relapsing polyneuropathy were studied by direct immunofluorescence. Granular deposits containing IgM and occasionally IgG were found in intraneural blood vessels and linear deposits of IgM were found on the Schwann cell plasmalemma of yet unmyelinated portions of nerve fibers. 59
In some patients, for example, antibodies to glycolipids LM1, GM1, or GD1b were detected, although less frequently than in GBS.61,62
Furthermore, a study showed that also P0, a protein responsible for myelin compaction, can be an autoantigen in some patients with CIDP. In this study, the sera of 21 CIDP patients were examined; six sera contained anti-P0 immunoglobulin G antibodies, and four of these caused conduction block and demyelination after intraneural injection in experimental animals. 63
Some studies, then, report that the incidence of CIDP is higher in patients with melanoma or after vaccination with melanoma lysates. 64 Interestingly, several carbohydrate epitopes, such as GM3, GM2, GD3 are shared both in myelin and in melanoma cells suggesting how molecular mimicry may be a possible trigger. 54
Recent studies, however, suggest that molecules not only in the compact myelin, but also within the nodal or paranodal regions may be probable targets of the immune attack in a minority of CIDP patients, but the literature pertaining about these proteins is still limited.65,66
Dysfunctions in these regions could explain the rapid changes in clinical symptoms seen in CIDP patients after treatment with PE or IVIg. In fact, while myelin disruption and axon loss are clear pathologic factors that lead to disability, 44 fast recovery after treatment cannot be explained on the basis of remyelination, but rather by functional conduction block induced by humoral factors against molecules at the NOR, region associated with saltatory conduction.5,6 Also, the worsening seen at the end of treatment effect, often predictable at 3–5 weeks, probably is not due to another demyelinating episode, but to the reappearance of the conduction block across the nodes. 54
Over the past decade, several autoantibodies against proteins of NOR (Neurofascin, NF 186/140) and paranodes (Contactin1, CNTN1; Neurofascin 1 NF155; paranodin (CASPR1) have been described.65,66
Myelinated nerve fibers consist of four domains: the node, paranode, juxtaparanode, and internode.
Recently, NF186 and NF140, antigens located at the NOR, were reported as the main targets of autoantibodies in five patients. 67 The paranodal region, at the margin of the NOR, is the site where myelin sheath contacts the axon enabling a high density of Na+ channels at the NOR and K+ channels at the juxta-paranodes. 68
IgG4 anti-CNTN1 antibodies selectively disrupt the binding of the CNTN1-CASPR1 complex to NF155, 69 a paranodal myelin constituent that interacts with CNTN1 to attach the paranodal loop to the axolemma. Loss of this attachment changes the nodal architecture and exposes Kþ channels in the juxtaparanodal region to limit saltatory conduction, causing conduction block and slowing. 70
In CIDP patients with autoantibodies against nodal or paranodal components, there is not a unique phenotype; for example, patients with anti-NF155 antibodies have younger ages of onset, disabling tremor, sensory ataxia, very high CSF protein levels, and poor response to IVIg. 71 The phenotype associated with CNTN1 antibodies is more variable, with some patients presenting at advanced ages with predominant motor involvement, rapid symptom onset, early axonal involvement, and poor response to IVIg, while others present with clinical features similar to anti-NF155. 66 Autoantibodies to nodal/paranodal proteins have been reported also in other forms of neuropathies such as multifocal motor neuropathy (MMN). 72
The broad phenotypic variability in the clinical spectrum of CIDP suggests that several mechanisms play a role in the immunopathological process. Although significant progress has been made, more studies are still needed to explain the complex immunopathogenesis of the majority of CIDP cases.
For example, differences in peripheral myelin antigen-specific T-cell responses and T memory subsets have been recently reported in atypical versus typical CIDP. 73 This evidence suggests that typical and atypical forms may probably subtend different pathogenetic mechanisms.
Diagnosis
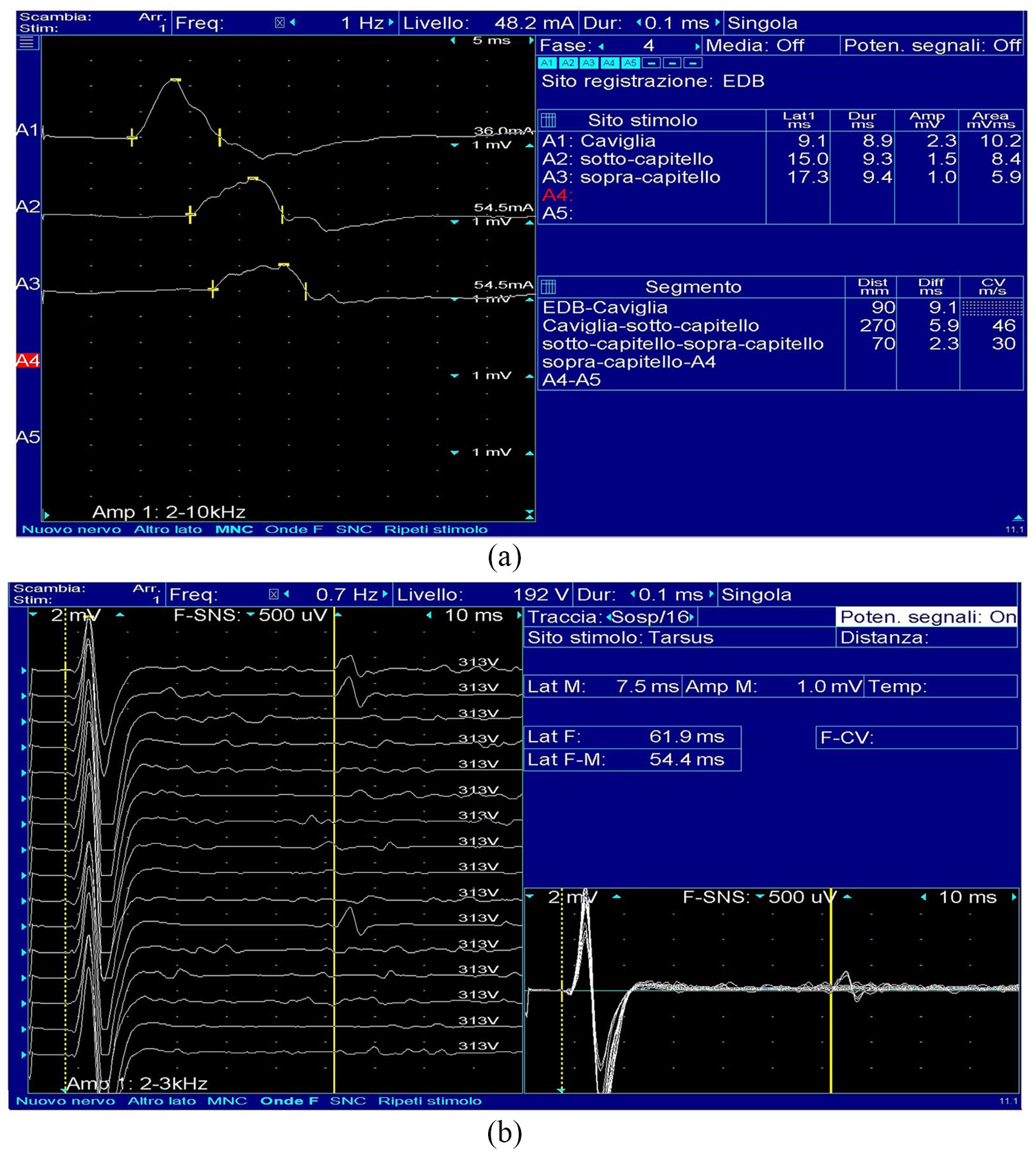
Diagnosis of CIDP is based on the documentation of demyelination on sensory and motor nerve conduction studies. Typical features are slowed motor conduction velocity, prolonged F-waves and distal motor latency, temporal dispersion of CMAP and motor conduction blocks in accord with the EFNS/ PNS criteria (Figure 2). 42

Nerve conduction studies in two patients affected by Chronic inflammatory demyelinating polyneuropathy. Electrodiagnostic studies performed in a CIDP patient; in (a) a proximal partial conduction block and severe temporal dispersion with polyphasic CMAP of the median nerve; in (b) a second CIDP patient presents reduced nerve conduction velocity, prolonged distal motor latency, temporal dispersion with polyphasic CMAP of the median nerve.
As in GBS, CSF shows albumin-cytologic dissociation with high CSF proteins and normal cell count. If pleocytosis is present, HIV, Lyme’s disease, sarcoidosis, and lymphoma should be considered as alternative diagnoses.74,75
Nerve biopsies may demonstrate “onion bulbs,” which are indicative of recurrent demyelination-remyelination, segmental demyelination, mononuclear cell infiltrates, Schwann cell proliferation and endoneurial collagen deposition (Figure 3).4,44
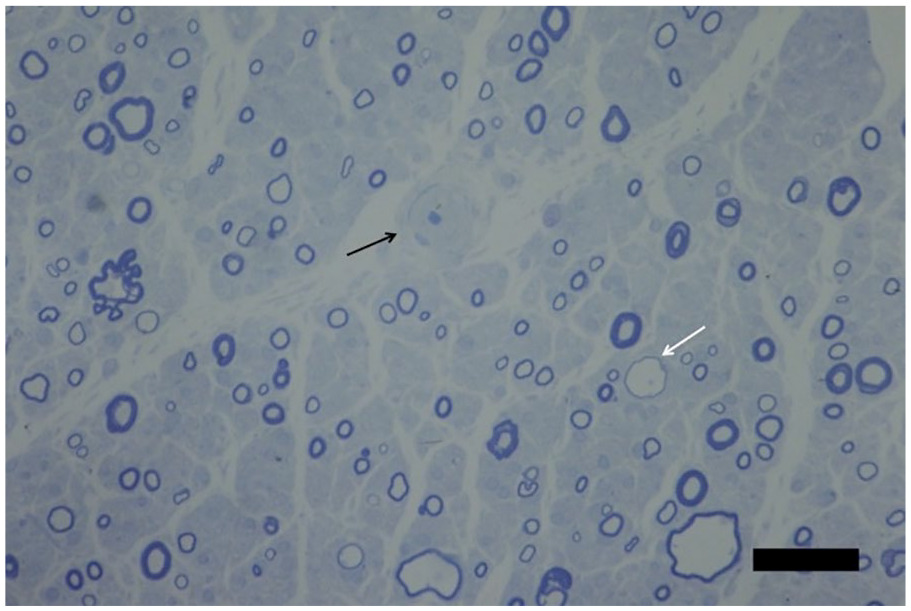
Nerve biopsy from a patient with CIDP. Semi-thin transverse section (0.7 μm) of sural nerve.
Treatment
Usual CIDP therapeutic options include corticosteroids, IVIg, PE and immunosuppressant drugs.
EFNS/ PNS guidelines, stated in 2010, recommend the use of IVIg (2 g/kg) or corticosteroids (prednisolone 60 mg/day) as first-line induction treatment, when disabling sensory and motor symptoms are present (while patients with mild symptoms, not interfering with activities of daily living, might be followed up without treatment). 42 PE, albeit equally effective, may have lower tolerability. When CIDP has pure motor involvement, only IVIg should be considered as initial treatment. 42 If a first-line treatment is ineffective or causes adverse events, other first-line therapies should be considered before passing to second-line strategies such as immunosuppressant drugs or combination approaches. For maintenance therapy, the continuation of the first-line approach is recommended until the maximum benefit is reached, with a subsequent tapering of the dose to determine the lowest effective amount. 42
Regarding the use of corticosteroids, even if broadly utilized, such guidelines have been called into question by recent meta-analyses.76,77 In particular, it was highlighted that only one trial compared oral prednisone, at a dosage of 120 mg every second day (and 5 mg on alternate days) tapering to 0 mg over 13 weeks, with no treatment. 78 In this study, the authors concluded that corticosteroids were significantly effective in reducing sensory-motor impairment and improving nerve conduction outcomes. Nonetheless, further analysis revealed that evidence from this study had a very low quality with a high risk of bias and incorrect results because many patients were excluded from the analysis as they infringed the protocol and only results for the remaining patients were reported. 76 No study compared corticosteroids to placebo. 76 One trial compared daily oral prednisolone with high-dose oral dexamethasone. There was no significant difference in reaching and keeping remission after 12 months, although there was a more rapid improvement in the dexamethasone group. 79
Also, corticosteroids have many and well-known side effects, whose incidence is higher in protracted treatments. Moreover, their occurrence may be at times caused even by low (⩽7.5 mg/d) dosages. Common side effects include osteoporosis, avascular necrosis of bones, myopathy, effects on glucose metabolism (mild increase in fasting and postprandial blood glucose levels), dyslipidemia, weight gain and cushingoid features, adrenal suppression, growth suppression, gastrointestinal effects (gastritis, peptic ulcer formation, visceral perforation, hepatic steatosis, pancreatitis, and gastrointestinal bleeding), hypertension, coronary heart disease, dermatoporosis, neuropsychiatric alterations (minor mood changes, depression, euphoria, mood lability, irritability, akathisia, and anxiety), cataract, glaucoma, and immunosuppression. 80 Therefore, corticosteroid administration should always be weighed, considering the potential risks and benefits for the patient.
The exact mechanism of action of IgG in CIDP is not fully understood, but the modulation of the FC region of IgG is hypothesized to play an important role. 31 The largest randomized placebo-controlled study was conducted on 117 patients and tested the efficacy of a baseline loading dose of 2 g/kg over 2–4 days followed by a maintenance infusion of 1 g/kg over 1–2 days every 3 weeks for up to 24 weeks versus placebo. 81 According to this study, patients treated with IVIg experienced an improvement in disability and strength, with a longer time to relapse. Two trials directly compared the efficacy of corticosteroids and IVIg,82,83 but there was little or no difference in a short-term improvement of disability between the two different treatments. 77
In 2018, the first randomized, double-blind, placebo-controlled trial on subcutaneous administration of Ig (SCIg), conducted on 172 patients, showed that SCIg were efficacious in preventing CIDP relapses and well-tolerated, suggesting its suitability as a maintenance treatment. 84
One trial compared IVIg with PE 85 and showed no significant short-term differences in change in impairment. PE has been proven to cause a clinically important improvement in disability in two trials;86,87 however, patients also experienced adverse events in 3%–17% of cases, related in difficulties with venous access, hypocalcemia related to the use of citrate and hemodynamic changes (mostly hypotension).
Concerning immunosuppressant drugs, only one randomized trial tested the efficacy of azathioprine (2 mg/kg) in addition to prednisone alone. 88 The quality of the evidence from this trial is very low; therefore, it is not possible to establish its efficacy in CIDP treatment. 77 One trial showed that there is little or no difference in change in disability scores between methotrexate (given in escalating doses from 7.5 to 15 mg weekly for 32 weeks) and placebo. 89 Similarly, a trial on interferon beta 1a did not show benefits in patients when compared to placebo. 77
Future directions
Evidence-based treatments for GBS and CIDP include IVIg and PE; moreover, in CIDP only, corticosteroids are commonly used in clinical practice. However, several unproven treatments are given in treatment-refractory disease.2,7
There is little evidence for many immunomodulating and immunosuppressive compounds, which have been proposed as therapeutic options, but randomized controlled trials are still needed. In the Table 1, we provide an overview of the current evidence on possible new treatments in GBS and CIDP and ongoing trials.
The table presents new possible therapeutic options in inflammatory neuropathies.

CIDP: chronic inflammatory demyelinating polyneuropathy; GBS: Guillain–Barré syndrome; RCT: randomized clinical trial.
Fingolimod, an immunomodulatory drug used in multiple sclerosis, constitutes a promising therapeutic candidate for CIDP. Recent data from studies in vitro suggested that fingolimod might induce nerve regeneration promoting Schwann cell phenotype. 90 A randomized clinical trial (RCT) of Fingolimod versus placebo in CIDP was performed and terminated prematurely due to clinical futility. 91
Alemtuzumab is an anti-CD52 monoclonal antibody used in the treatment of chronic lymphocytic leukemia and multiple sclerosis. 92 In a small cohort of patients, it was reported a beneficial effect in prolonged remission in young patients with short disease duration.93,94 However, its larger application is still limited because of many side effects, such as autoimmune thyroiditis.
Natalizumab acts against cellular adhesion and T-cell migration by direct action on the α4β1 integrin. It is used in multiple sclerosis and was initially described as producing deterioration in a single patient with CIDP. 95 Treatment of three subjects at a single center resulted in inconsistent outcomes. 96
Etanercept is a tumor necrosis factor-α antagonist. It is used effectively in rheumatoid and psoriatic arthritis. It was used in 10 reported patients with CIDP with promising effects. 97 However, Etanercept has been associated with central and peripheral demyelination. 98
Rituximab is an anti-CD20 monoclonal antibody that has been used successfully in neuromuscular diseases and in a small number of patients with CIDP. 99 , 100 Several reports and case series describe the efficacy of rituximab in CIDP, and it was proposed in refractory disease. 100 ,101 Further anti-CD20 drugs, such as ocrelizumab and ofatumumab, might constitute future therapeutic options.
Based on the potential to suppress complement-mediated nerve damage, a phase 2 study was conducted to assess the safety and efficacy of the administration of 900 mg of eculizumab (a humanized monoclonal antibody against the complement protein C5) plus IVIg in subjects with severe GBS. 102 Although eculizumab did not reach the expected primary outcome in that study on GBS, this treatment had good results in patients with a mutation in the CD59 gene and the murine model of GBS,103,104 thus remaining a promising treatment requiring further investigation. 105
IFN-β-1a is a cytokine which downregulates inflammatory responses, used for relapsing-remitting multiple sclerosis. A few reports suggested a possible effect in GBS, but two RCTs failed to confirm any benefit in CIDP.106,107
Bregs and Tregs have been implicated in modulating CIDP pathogenesis and disease progression based on new perspectives derived from murine SAP models; hence, therapies aiming at the expansion of Bregs and Tregs may be an effective approach in autoimmune neuropathies. 49
Ongoing and future trials may point out additional therapies in CIDP. However, newer agents require adequate double-blind, placebo-controlled, randomized trials to improve our understanding of their potential benefits and toxicities. Homogeneous populations supported by collaborative research to better define inflammatory polyradiculoneuropathies, guided by the use of peripheral nerve and nerve root specimens, CSF or blood from these patients should further aid our understanding of these disorders using uniformly collected data. There is a need to compare responsiveness to different treatments based on valid and reliable measures and standardize their use in evaluating patients with inflammatory polyradiculoneuropathies. Automated, non-biased high throughput screening approaches (e.g. transcriptomics, proteomics) can offer more reliable data to study patients affected by inflammatory polyradiculoneuropathies compared to age- and sex-matched non-inflammatory neuropathy patients and healthy controls. Changes in levels of autoantibodies should also be monitored closely and more efforts should be done to identify predictors of response to molecularly targeted agents. Finally, adequate randomization and study of CIDP variants in clinical trials are required.
Limitations
The principal limitation of this study is that it is a narrative review and not a systematic review or a meta-analysis. However, the aim of the study was not to present an evidence-based review of the sparse literature on inflammatory polyradiculoneuropathies but to make the reader aware of the new perspectives and advance in the pathophysiology and mechanisms of these diseases with an update on diagnosis and therapy. The majority of the studies analyzed presented many pitfalls that could have made difficult to evaluate the efficacy of different treatment regimens in GBS and CIDP; first of all, the measures of outcome and the grade of involvement among different studies are heterogeneous. Although a well-documented efficacy is available to date for some clinical instruments, less powerful evidence is available for others. A further limitation emerges from the inhomogeneity in the treatment in patients treated with immunosuppressants, monoclonal antibodies (i.e. Rituximab), and steroids. It is a well-known fact that steroids can impact on the severity of the disease and this issue might have influenced the interpretation of results from the mentioned studies.
Conclusion
Inflammatory polyradiculoneuropathies are disorders with undetermined pathogenesis without specific therapies. However, our knowledge on the immunopathogenesis is still evolving and significant efforts have been made in the last years to understand their pathogenesis.
Little is known about the determinants of susceptibility in inflammatory neuropathies, but many studies in animal models, as well as in patients with inflammatory neuropathies are ongoing.
Diagnosis is moreover clinical, but nerve conduction studies and MRI have an important role in the differential diagnosis. Evidence-based treatments for GBS and CIDP include IVIg and PE. In CIDP only, corticosteroids are frequently used with a good efficacy. These treatments are effective in typical forms when started early from disease onset, but there is a lack of efficacy in clinical variants and more specific and target therapies are still needed.
Our understanding of disease mechanisms in inflammatory neuropathies has improved in the last years. Furthermore, recent advances in immunology and molecular biology are going to arrive in the pathogenesis of these disorders and new target antigens will probably be described in the next future. Several drugs might be candidates for possible use in clinical practice, but newer agents require adequate randomized controlled trials to demonstrate their efficacy and safety profiles.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Informed consent
The authors have obtained the necessary permission directly from the patients to reproduce the figures in this article (biopsies and nerve conduction studies).