
Abstract
Objectives
Alantolactone (AL) is a compound extracted from the roots of Inula Racemosa that has shown beneficial effects in cardiovascular disease. However, the cardioprotective mechanism of AL against hypoxic/ischemic (H/I) injury is still unclear. This research aimed to determine AL’s ability to protect the heart against isoproterenol (ISO)-induced MI injury in vivo and cobalt chloride (CoCl2) induced H/I injury in vitro.
Methods
Electrocardiography (ECG), lactate dehydrogenase (LDH), creatine kinase (CK), and cardiac troponin I (cTnI) assays in addition to histological analysis of the myocardium were used to investigate the effects of AL in vivo. Influences of AL on L-type Ca2+ current (ICa-L) in isolated rat myocytes were observed by the patch-clamp technique. Furthermore, cell viability, apoptosis, oxidative stress injury, mitochondrial membrane potential, and intracellular Ca2+ concentration were examined in vitro.
Results
The results indicated that AL treatment ameliorated the morphological and ECG changes associated with MI, and decreased levels of LDH, CK, and cTnI. Furthermore, pretreatment with AL elevated antioxidant enzyme activity and suppressed ROS production. AL prevented H/I-induced apoptosis, mitochondria damage, and calcium overload while reducing ICa-L in a concentration and time dependent fashion. The 50% inhibiting concentration (IC50) and maximal inhibitory effect (Emax) of AL were 17.29 μmol/L and 57.73 ± 1.05%, respectively.
Conclusion
AL attenuated MI-related injury by reducing oxidative stress, apoptosis, calcium overload, and mitochondria damage. These cardioprotective effects may be related to the direct inhibition of ICa-L.
Introduction
Myocardial ischemia (MI) is one of the cardinal pathological features of ischemic heart disease, which is the leading cause of death worldwide. 1 MI involves reduced blood perfusion of the heart, resulting in decreased oxygenation and aberrant energy metabolism that are insufficient to support healthy cardiac function. 2 Prolonged ischemia can lead to a point beyond which the cellular damages become irreversible and apoptosis becomes inevitable. Both oxidative stress and intracellular calcium overload have been implicated in MI.3,4 The synergistic effects of oxidative stress and calcium overload induce cardiac cell dysfunction and apoptosis, which drastically impairs cardiac function. 5
When the heart is subjected to hypoxia/ischemia (H/I), the concentration of calcium ions (Ca2+) increases. An excessive increase in the intracellular calcium concentration would result in calcium overload and thus produce myocardial cell injury.6,7 The Na+/Ca2+ exchanger (NCX) is central to the mechanism of calcium efflux from cardiac myocytes. 8 Calcium is pumped out of the myocardial cell largely by the sarcolemmal NCX with some contribution from the sarcolemmal Ca-ATPase (PMCA). 9 These Ca2+ are located on the sarcoplasmic reticulum (SR) and the release of Ca2+ from the SR is termed “calcium-induced calcium-release.” 10 In cardiomyocytes, L-type Ca2+ channels (LTCCs) are primarily responsible for the influx of Ca2+, an essential intracellular messenger associated with the entire life cycle of this specialized cell type. 11 Inhibiting LTCCs results in reduced Ca2+ entry, and thus diminishes any Ca2+ overload. Reestablishing this Ca2+ homeostasis has a cardioprotective effect against MI injury. 12
Augmented oxidative stress is one of the major elements of the complex pathophysiological mechanism underlying MI. 13 Oxidative stress results from the increased formation of reactive oxygen species (ROS) and causes DNA damage, protein and lipid peroxidation, and cellular dysfunction, all of which are associated with the pathological injury of the heart. 14 In most cell types, mitochondria are the primary source of ROS, 15 yet the damage ROS cause is not restricted to mitochondrial macromolecules but extends to the surrounding intracellular space as well. 16 Furthermore, ROS and mitochondria play important roles in apoptosis induction under both physiological and pathological conditions. 17 The most prevalent ROS include the peroxynitrite anion (ONOO–), the superoxide anion radical (dioxide or O2–), and the hydroxyl radical (OH). Their high reactivity causes cellular dysfunction and sometimes cells death. 18 Myocardial injury causes excess production of ROS, and this oxidative stress leads to mitochondrial dysfunction and further aggravates cardiomyocyte apoptosis. 19 In the heart, mitochondria supply more than 90% of the adenosine triphosphate (ATP) required by the heart through oxidative phosphorylation. 20 Consequently, mitochondrial damage in MI due to oxidative stress causes excessive ROS production, dysfunction of mitochondria, and cellular injury.
Currently, natural products are an important resource for the development of small molecules for the treatment of various disease.
21
Alantolactone (AL, Desit, China; Figure 1) is a compound extracted from the roots of Inula Racemosa that exhibits a diversity of biological activities including anti-inflammatory, anti-cancer, anti-bacterial, and anti-fungal effects.
22
Inula Racemosa protects the heart from ISO-induced myocardial injury by reducing oxidative stress and modulating the hemodynamic and ventricular functions of heart.
23
In addition, Inula Racemosa exerts cardioprotective effects in ischemic rats and protects the myocardium of the rat heart from oxidative damage. However, the protective mechanism of AL in ischemic myocardial injury remains unknown. Chemical structure of alantolactone. (Molecular formula: C15H20O2; Molecular weight, 232.32).
Isoproterenol (ISO), a synthetic catecholamine, induces MI by increasing the force and frequency of myocardial contractions and raising intracellular Ca2+ concentration. 24 The ISO-induced MI model is not directly analogous to human occlusive MI, and this model may be more pertinent to MI which may involve excessive compensatory over-activity of the sympathetic nervous system. 25 The ISO-induced MI model is one of the most widely used experimental models to study the beneficial effects and cardiac functions of various drugs. 26 Cobalt chloride (CoCl2) is a hypoxia-mimicking agent because of its property to mimic hypoxic/ischemic (H/I) conditions. 27 To model MI in vivo and H/I in vitro, in the present study, rats were injected with ISO and H9c2 cells were exposed to CoCl2, respectively. The present study aimed to determine whether AL can play a protective role in the heart by reducing oxidative stress and regulating Ca2+ homeostasis.
Materials and methods
Experimental animals and treatment
Adult male Sprague–Dawley (SD) rats (weigh: 200 ± 20 g; age: 8 weeks old) were supplied by the Laboratory Animal Center of Hebei Medical University. The rats were housed at 25 ± 1°C with 55 ± 5% humidity, on a 12 h light-dark cycle, and supplied with food and water. All procedures were carried out under the Guidelines of Animal Experiments from the Committee of Medical Ethics and approved by the Ethics Committee for Animal Experiments of Hebei University of Chinese Medicine (approval number: DWLL2020073).
Forty rats were evenly and randomly divided into four treatment groups (n = 10 rats per group): control (CON), isoprenaline (ISO), low dose AL group (L-AL), and high dose AL group (H-AL). The L-AL and H-AL groups received 25 and 50 mg/kg/d by oral gavage, respectively. Rats in the ISO and CON groups were gavaged with an equal volume of saline daily. After 7 days of continuous treatment, the rats in all but the CON groups were administered ISO (85 mg/kg) by subcutaneous injection on two consecutive days. After the final injection on day 9, the rats were deprived of food and water for 24 h. On day 10, electrocardiograms (ECGs) were performed, blood samples were collected, and the hearts were rapidly removed after administering anesthesia (sodium pentobarbital, 50 mg/kg according to body weight).
Detection of electrocardiogram (ECG), cardiac functional parameters, and cardiac marker enzymes
At the end of the experimental period, a standard limb leads II ECG was performed in rats. ECG recordings were obtained from the rats by using BL-420S experimental system. Catheterization of the left ventricle (LV) was performed on anesthetized rats after the final injection of ISO. A miniature pressure transducer was inserted into the aorta via the right carotid artery and advanced into the LV under continuous monitoring of the pressure waveform. LV systolic pressure (LVSP), LV end-diastolic pressure (LVEDP), and ± dp/dtmax were monitored continuously and then recorded and analyzed after 10 min of stabilization.
Sera were obtained by centrifuging the whole-blood samples. The levels of lactate dehydrogenase (LDH), creatine kinase (CK), and cardiac troponin I (cTnI) were measured using commercial kits according to the manufacturer’s protocol (Nanjing Jiancheng Institute, China).
Histopathological examination of heart tissues
Heart tissue samples were fixed in 4% paraformaldehyde hydrated for 24 h before being embedded in paraffin. After standard hematoxylin and eosin (HE) staining, the sections of mid-myocardium (4 μm thick) were examined under a light microscope.
Immunohistochemistry
The tissue sections were dewaxed, rehydrated, and immersed in retrieval solution. Subsequently, the sections were treated with 3% hydrogen peroxide and incubated at 25°C for 20 min, and then washed in phosphate buffer solution (PBS) 3 times for 5 min each. Any non-specific staining was blocked with 5% Ig blocking reagent and 5% serum (Shanghai Regal Biological Technology Development Co., Ltd.) for 15 min at 37°C after rinsing. Slides were incubated with diluted rabbit polyclonal antibodies against TNF-α and IL-6 at 4°C overnight. The next day, the slides were rinsed 3 times with PBS for 5 min each. Next, the slides were incubated with their corresponding secondary antibodies for 20 min at room temperature. The target protein was then stained with 3,30-diaminobenzidine tetrahydrochloride.
Cell culture and experiment groups
H9c2 cells were cultured in high glucose Dulbecco’s Modifed Eagle’s Medium (DMEM, Gibco) supplemented with fotal bovine serum (FBS, Gibco) and antibiotics (1% penicillin-streptomycin, Gibco) in an atmosphere of 95% air and 5% CO2 at 37°C. The medium was changed every 2–3 days. To select the appropriate concentration of AL, H9c2 cardiomyocytes were pretreated with AL at a range of concentrations (2.5, 5, 10, 20, and 40 μmol/L) for 24 h in untreated and CoCl2-induced H9c2 cells. The cell viability assays suggested that AL had the most obvious survival pro-survival effect at about 10 μmol/L. Consequently, 5 and 10 μmol/L were chosen as the low and high doses for all subsequent experiments. The cells were classified into four groups: (1) CON (blank control of H9c2 cells); (2) CoCl2 (H9c2 cells incubation with CoCl2 (600 μmol/L)); (3) L-AL (H9c2 cells incubatd with CoCl2 (600 μmol/L) and AL (5 μmol/L)); and (4) H-AL (H9c2 cells incubatd with CoCl2 (600 μmol/L) and AL (10 μmol/L)). H9c2 myocytes in the CON group were cultured under normoxic conditions. The cells in the CoCl2 group were cultured with 600 μmol/L CoCl2 for 24 h. The cells in the L-AL and H-AL groups were pretreated with 5 and 10 μmol/L AL for 24 h, respectively, followed by incubation with 600 μmol/L CoCl2 for 24 h.
Detection of cell viability
The effect of AL on cell viability was evaluated using the Cell Counting Kit-8 (CCK-8, Biosharp, China) assay. H9c2 cells were seeded in 96 well cell culture plates 1 × 104 cells/well. After the aforementioned treatments, a solution of 10 μL of CCK-8 diluted in 100 μL DMEM was added to each incubated for 2 h at 37°C. The OD at 450 nm was measured using a microplate reader (Thermo, USA).
Detection of LDH and CK release in culture medium
Cellular injury was assessed by LDH and CK release. H9c2 cells were cultured at 1 × 106 cells/well in six well plates. The cell culture medium was collected after the cell viability experiment (described above). The contents of LDH and CK were quantified using commercial kits according to the manufacturer’s instructions (Nanjing Jiancheng Institute, China).
Evaluation of cell apoptosis using Hoechst-33258 staining and flow cytometry
Following treatment, the cells from each group were washed with cold PBS and incubated with Hoechst 33342 dye for 15 min in the dark at room temperature. Hoechst fluorescence was visualized and imaged using a fluorescence microscope (Olympus, Japan).
The rate of apoptosis among H9c2 cardiomyoblasts was detected using Annexin V-FITC/PI Apoptosis Kit in accordance with the manufacturer’s instructions. The cells from the four treatment groups were collected and washed 3 times with PBS before being resuspended in a prepared binding buffer (100 μL). The cell suspensions were transferred into a 5 mL flow tube and stained with 5 μL Annexin V-FITC/PI that incubate at room temperature for 15 min in the dark. Finally, the cells were analyzed using a flow cytometer.
Measurement of intracellular superoxide dismutase (SOD), catalase (CAT), glutathione (GSH), and malondialdehyde (MDA)
H9c2 cells were cultured at a density of 1 × 106 cells/well in six well plates. Then, H9c2 cells were washed with cold PBS, and centrifuged at 14,000 r/5 min. The supernatant was removed and the precipitate was sonicated and the H9c2 cell lysate was resuspended. The activities of CAT, SOD, MDA, and GSH were analyzed using commercial kits (Nanjing Jiancheng Institute, China).
Measurement of intracellular ROS production
ROS production was assessed with the peroxide-sensitive fluorescent probe 2′,7′-dichlorofluorescin in diacetate (DCFH-DA). The dye was loaded by incubating the H9c2 cells with 20 μmol/L DCFH-DA for 15–20 min at 37°C. The cells were then visualized imaged using a fluorescence microscope (Olympus, Japan).
Measurement of Mitochondrial membrane potential (MMP)
Rhodamine 123 was used to estimate the electrical potential across the inner mitochondrial membrane. Following the experimental treatments, H9c2 cells were washed with PBS and incubated with rhodamine 123 for 15 min at 37°C. Subsequently, the cells were washed with PBS twice, and images were captured with a fluorescence microscope (Olympus, Japan).
Determination of intracellular Ca2+ concentration
To determine the intracellular Ca2+ concentration, H9c2 cardiomyocytes were incubated with Fluo-3/AM in the dark for 20 min. Then, the cells were visualized and imaged by using a fluorescence microscope (Olympus, Japan).
Isolation of rat ventricular myocytes
To obtain normal cardiomyocytes, rats were injected with heparin (500 IU/kg) intraperitoneally and then anesthetized with ethyl carbamate (1.0 g/kg) 20 min later. The hearts were then rapidly excised and perfused for 5 min at a rate of 4 mL/min via the aorta with oxygenated ice-cold Ca2+-free Tyrode’s solution (NaH2PO4 0.33 m
To obtain ischemic cardiomyocytes, rats were injected with ISO subcutaneously (85 mg/kg). After treatment with ISO, the hearts were excised as described above to isolate the cardiomyocytes.
Electrophysiology
The Ca2+ current was detected in rat ventricular myocytes using a whole-cell patch-clamp technique. The whole-cell configuration was maintained at a room temperature using a glass pipette with a tip resistance of 2–5 MΩ filled with pipette solution (TTX 10 μ
For all experiments, the holding potential was −80 mV followed by depolarization to 0 mV. Meanwhile, the Ca2+ currents were elicited by a 200 msec depolarizing pulse. Membrane capacitance (Cm) was estimated from capacitive transients and calculated according to Cm = τm × IoVm (1 − Iss/Io), where τm is the membrane time constant, Io is the maximal amplitude of the capacitive current spike, Iss is the current at the end of the 10 msec pulse (steady state), and Vm is the amplitude of the voltage step (Cm = 152.8 ± 23.7 pF). Axon patch 200B amplifier and pClamp 10.2 software (Axon Instruments, Union City, CA, USA) were used to record the currents.
Statistical analysis
All data were analyzed and fitted b using Origin 7.5 (OriginLab Corp., Northampton, USA) and Clampfit 10.2 software (Molecular Devices, Sunnyvale, USA) software. The inhibition ratio of AL on ICa-L was calculated (IControl – IDrug)/IControl. The concentration-response curve was fit with the logistic equation: y = A2 + (A1 – A2)/[1 + (x/x0) p ], where p is the Hill coefficient, A1 is the maximum responses, A2 is the minimum responses, and x is the drug concentration and y is the response. The steady-state activation and inactivation curves of ICa-L were fit with Boltzmann functions: y = A/{1 + exp [(Vh – Vm)/k]}, where A is the amplitude of the relationship, k is the slope, Vm is the test potential, and Vh is the voltage at half-maximal activation.
All data are presented as mean ± standard error of the mean (SEM). Comparisons were made via one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test. p < 0.05 was considered reflective of statistical significance.
Results
Effects of AL on ECG, cardiac functional parameters, and cardiac marker enzymes
The needle electrode was inserted subcutaneously between the paw pads of each rat and the ECG was recorded continuously. Compared with the CON group, the ST interval increased, and the amplitude of the R wave decreased in the ISO-induced group (Figure 2(a)–(c)). The L-AL and H-AL groups showed ST interval and R wave amplitude recovery. As shown in Table 1, ISO treatment caused significant increases (p < 0.01) in LVEDP compared with the CON group. On the other hand, ISO caused significant decreases in LVSP and ± dp/dtmax compared to the CON group. Table 1 shows that AL pretreatment restored both parameters to nearly baseline levels (p < 0.01). Effects of AL on ECG, histopathology, and cardiac enzymes. (a) Representative ECG tracings. (b and c) Statistical analysis of heart rate and J-point elevation. (d) Representative microscopic photographs of hearts stained with H&E (magnification: ×400, Scale bar: 50 μm). Those arrows distinguish clearly for edema (red arrow), inflammatory cell infiltration (green arrow), and either pyknotic or deepening nucleus (black arrow). (e–g) cTnI, LDH and CK levels in serum. The values are the means ± SEM (n = 10, ##p < 0.01 vs CON; **p < 0.01 vs ISO). Effects of AL on changes of cardiac functional parameters. The values are the means ± SEM (#p < 0.05 vs CON; ##p < 0.01 vs CON; *p < 0.05 vs ISO; **p < 0.01 vs ISO).
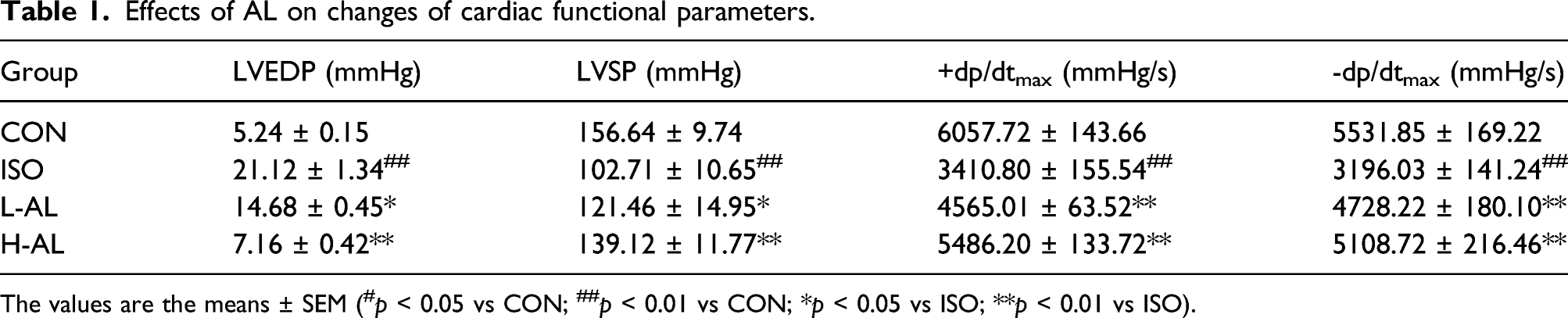
As shown in Figure 2(e)–(g), the levels of LDH, CK, and cTnI, heart rate, and J-point elevation were increased in the ISO group, indicated that the model of MI in this research was successfully established (p < 0.01). Compared with the ISO group, J-point elevation, heart rate, and LDH, CK, and cTnI levels in the L-AL and H-AL groups were decreased (p < 0.01).
Effects of AL on histopathology
The myocardial tissue structure in the CON group was clear, myocardial cell arrangements were orderly, the cell membrane was intact, the nucleus was clear, and no pathological change was observed. As shown in Figure 2(d), the heart tissues from ISO-induced MI rats exhibited obvious interstitial edema, inflammatory cell infiltration and either pyknotic or deepening nucleus, the results are consistent with previous study. 28 In contrast, the L-AL and H-AL groups showed clear transverse striations, indicating that pretreatment with AL suppressed the ISO-induced myocardial pathology.
Effects of AL on IL-6 and TNF-α expression levels
The expression levels of IL-6 and TNF-α in cardiac tissue were assessed using immunohistochemistry. As shown in Figure 3, IL-6 and TNF-α expression were low in the CON group and increased after ISO treatment. AL pretreatment reversed these inflammatory expressions. The expression of inflammation in L-AL and H-AL groups was significantly lower than that in the ISO group. Effects of AL on IL-6 and TNF-α expression as detected by immunohistochemistry. Heart tissues were obtained from the CON, ISO, L-AL, and H-AL groups. The morphological location and area percentages of IL-6 expression (a and c), and TNF-α expression (b and d) are shown. Magnification: ×400, scale bar: 50 μm. Positive expression of IL-6, and TNF-α is shown by arrows. The values are the means ± SEM (##p < 0.01 vs CON; *p < 0.05 vs ISO; **p < 0.01 vs ISO).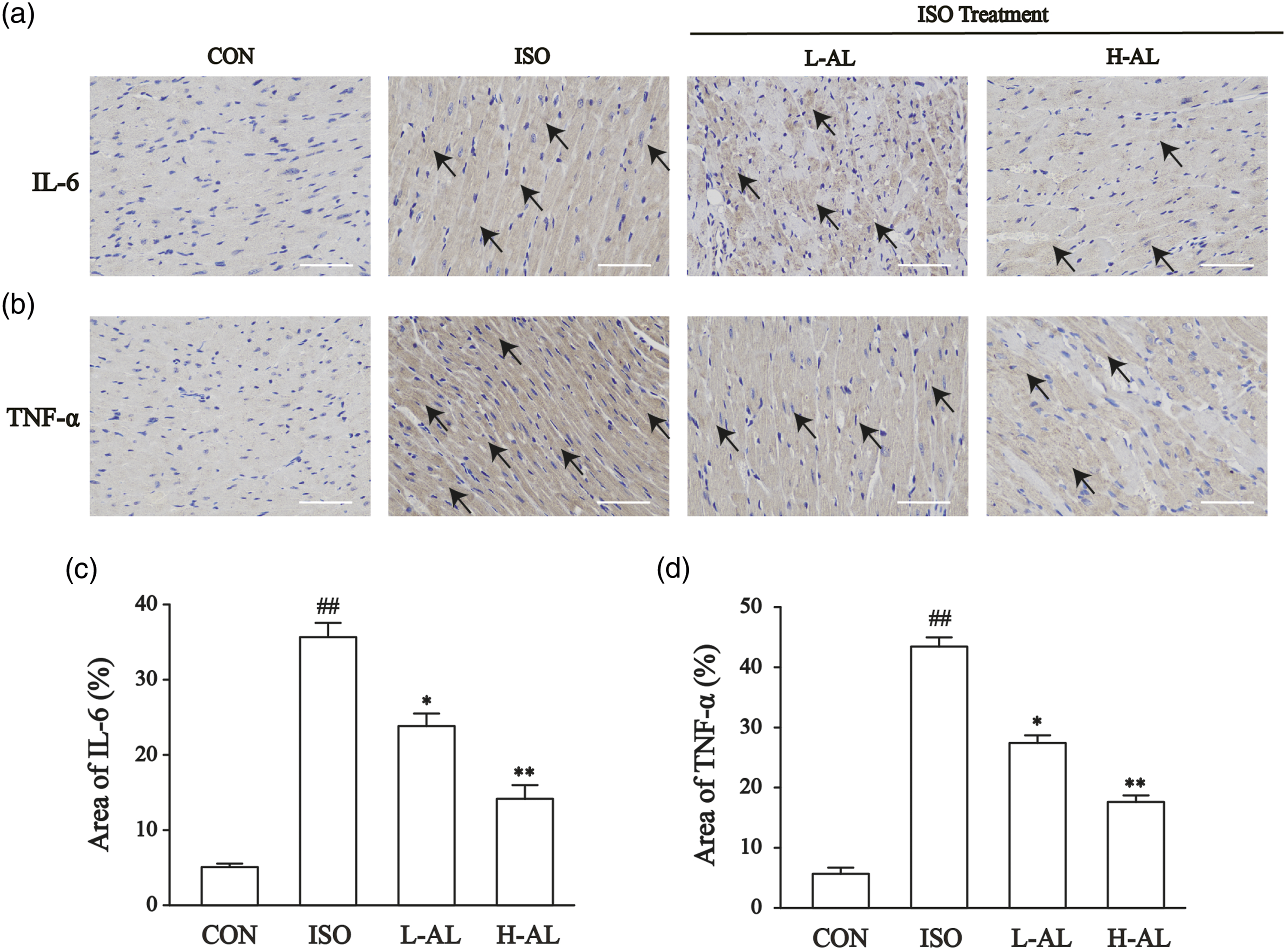
Effects of AL on cell viability
We analyzed the viability of H9c2 cells treated with different concentrations of AL using the CCK-8 assay (Figure 4). Incubating H9c2 cells with increasing concentrations of CoCl2 (400–800 μmol/L) significantly reduced their viability (p < 0.01, p < 0.05). Figure 4(b) displayed that 2.5, 5, 10, and 20 μmol/L AL treatment not significant effect on H9c2 cell viability. AL concentrations of 5 and 10 μmol/L were selected for all further experiments. Compared with CON group, exposure to CoCl2 for 24 h reduced the growth of H9c2 cells (p < 0.01, p < 0.05). However, pretreatment with AL significantly increased the viability of H9c2 cells (p < 0.01). Effects of AL on H9c2 cell viability. (a) Cell viability was measured following treatment with CoCl2 (0–800 μmol/L). (b) Cell viability of H9c2 cell was measured following treatment with AL (2.5, 5, 10, 20 and 40 μmol/L). (c and d) Cell viability of H9c2 cell was measured following treatment with AL and CoCl2. The values are the means ± SEM (n = 6, #p < 0.05 vs CON; ##p < 0.01 vs CON; *p < 0.05 vs CoCl2; **p < 0.01 vs CoCl2).
Effects of AL on LDH and CK activities in culture medium
As shown in Figure 5(c) and (d), the enzymatic activities of LDH and CK increased in the CoCl2 group compared with the CON group (p < 0.01). The activities of LDH and CK present in the culture medium decreased when AL pretreatment preceded the addition of CoCl2 (p < 0.01, p < 0.05). Effects of AL on apoptosis and cardiac marker enzymes. (a and b) The morphological changes of H9c2 cells are shown by Hoechst-33258 staining (magnification: ×200, Scale bar: 100 μm). (c and d) LDH and CK levels in cell culture supernatant. (e and f) Apoptosis rates were determined using flow cytometry. The values are the means ± SEM (n = 6, ##p < 0.01 vs CON; *p < 0.05 vs CoCl2. **p<0.01 vs CoCl2).
Effects of AL on apoptosis
Compared with the CON group, apoptosis among the cells in the CoCl2 group had dramatically increased (p < 0.01). After being treated with CoCl2 for 24 h, the H9c2 cells presented with condensed and fragmented nuclei and apoptotic bodies (Figure 5(a) and (b)). Pretreatment with 5 and 10 μmol/L AL for 24 h before CoCl2 addition reduced the number of apoptotic cells significantly (p < 0.01).
Figure 5e shows the effects of AL on apoptosis as detected by flow cytometry. These results evidenced that the apoptosis rate of the CoCl2 group was significantly increased relative to the CON group. However, the apoptosis rate of the AL treatment group was significantly lower than the CoCl2 group (p < 0.01).
Effects of AL on oxidative stress
DCFH-DA was employed to evaluate ROS levels in H9c2 cells.
29
As shown in Figure 6(a) and (b), in contrast to the CON group, the amount of ROS in the CoCl2 group increased significantly (p < 0.01). Pretreatment with AL decreased intracellular ROS generation in H/I-induced cardiomyocytes (p < 0.01). These findings suggested that AL pretreatment prevents CoCl2-induced oxidative stress in cardiomyocytes. As shown in Figure 6(c)–(f), CoCl2 caused significant decreases in the activities of CAT, GSH and SOD compared with the CON group (p < 0.01). Pretreatment with AL significantly increased the levels of CAT, GSH, and SOD compared with the CoCl2 group (p < 0.01). Additionally, treatment with CoCl2 along increased the MDA concentration relative to control, and AL pretreatment prevented this effect (p < 0.01). Effects of AL on CoCl2-induced oxidative stress in H9c2 cells. (a and b) Intracellular oxidant stress evaluation with 2′,7′-dichlorofluorescin diacetate (DCFH-DA) labeling in H9c2 cells (magnification: ×200, Scale bar: 100 μm). (c–f) MDA content, and the activities of SOD, CAT, and GSH were measured in H9c2 cardiomyocytes. The values are the means ± SEM (n = 6, ##p < 0.01 vs CON; **p < 0.01 vs CoCl2).
Effects of AL on mitochondrial membrane potential (MMP)
The change of rhodamine 123 fluorescence to examine whether preservation of MMP is associated with cardioprotective effects of AL was assessed. The MMP in the CoCl2 group was downregulate compared with the CON group (p < 0.01). Meanwhile, the MMP was elevated significantly in the L-AL and H-AL groups, in contrast to the CoCl2 group (Figure 7(a) and (c)) (p < 0.01, p < 0.05). Effects of AL on mitochondrial membrane potential and Ca2+ concentration in H9c2 cells. (a) Representative image of rhodamine 123 fluorescence intensity in H9c2 cells (magnification: ×200, Scale bar: 100 μm). (b) Representative image of intracellular Ca2+ concentration fluorescence intensity in H9c2 cells (magnification: ×400, Scale bar: 50 μm). (c and d) Statistical analysis of rhodamine 123 and intracellular Ca2+ concentration fluorescence intensities, respectively. The values are the means ± SEM (n = 6, ##p < 0.01 vs CON; **p < 0.01 vs CoCl2).
Effects of AL on Ca2+ concentration
To determine the intracellular Ca2+ concentration of H9c2 cells, we quantified the ratio of Fura-3/AM staining. The result indicated that intracellular Ca2+ accumulation was increased in CoCl2-treated H9c2 cells (p < 0.01). The intensity of Ca2+ fluorescence dramatically decreased in H9c2 cells after pretreatment with 5 and 10 μmol/L AL (Figure 7(b) and (d)) (p < 0.01).
Confirmation of ICa-L
As shown in Figure 8(a) and (b), as a specific T-type Ca2+ channel blocker, NiCl2 (0.01 mmol/L) did not affect the inward currents, which demonstrated that the induced currents were not mediated by T-type Ca2+ channels. VER (1 μmol/L) is a specific LTCC blocker that attenuated the current almost entirely, indicating that these currents were ICa-L. Confirmation of ICa-L in myocardial cells. (a) Representative trace recorded after treatment with NiCl2 (0.01 m
Effects of AL on ICa-L
Figure 9(a)–(f) shows the reversible Ca2+ current recording after the effective dose of 10 μmol/L AL was added both in healthy and ischemic cardiomyocytes. AL reduced ICa-L with an inhibition rate of 42.62 ± 5.18% and 44.14 ± 4.56%, respectively. After washing out with external solution, the currents partially recovered. These results speak to the reversibility of AL’s effects on ICa-L. The reversible effect of AL on ICa-L. (a–c) Healthy and (d–f) ischemic ventricular myocytes in rats. (g) Exemplary traces and (h) time course of ICa-L were recorded under control conditions during exposure to 0.3, 1, 3, 10, and 30 μ
Concentration-dependent effects of AL on ICa-L
Figure 9(g)–(i) contains representative traces for a range of AL concentrations. With increasing concentrations of AL (from 0.3 to 30 μmol/L), ICa-L was gradually suppressed. The peak amplitude of ICa-L was reduced by 2.25 ± 0.17%, 12.70 ± 0.71%, 22.91 ± 1.12%, 42.78 ± 1.06%, and 57.73 ± 1.05% by AL derivatives at 0.3, 1, 3, 10, and 30 μmol/L, respectively, and the 50% inhibiting concentration (IC50) of AL was 17.29 μmol/L. Effects of AL on the current–voltage (I–V) relationship of ICa-L.
As shown in Figure 10(a) and (b), the effects of AL (3 μmol/L and 10 μmol/L) on the I–V relationship are depicted. The I–V curves showed an upward trend. The ICa-L amplitude increased from −20 mV and attained a maximum at 0–10 mV. These results demonstrated that AL 3 and 10 μmol/L reduced the maximum current by a significant margin. Effects of AL on the dynamical properties of the Ca2+ channel. (a) Representative ICa-L in the CON, AL (3, 10 μ
Effects of AL on steady-state activation and inactivation of ICa-L
Figure 10(c) and (d) shows that both concentrations of AL (3 and 10 μmol/L) affected the steady-state activation and inactivation of ICa-L. The mean half-maximum activation voltage (V1/2) value of activated ICa-L in the CON group was –11.60 ± 0.66 mV, and the slope factor (k) was 4.56 ± 0.62 mV. However, compared with the CON group, the values of V1/2 for activation with 3 μmol/L of AL was –12.84 ± 0.47 mV and the k value was 4.23 ± 0.40 mV. The values of V1/2 for activation with 10 μmol/L of AL were −13.45 ± 0.16 mV and the k value was 4.59 ± 0.13 mV. Without AL, V1/2 of inactivation was –24.30 ± 0.47 mV and the k value was 4.93 ± 0.39 mV. In the presence of 3 and 10 μmol/L AL, the values of V1/2 for inactivation were –26.95 ± 0.28 mV and –29.99 ± 0.57 mV, respectively, and the k values were 3.69 ± 0.22 mV and 4.37 ± 0.60 mV, respectively.
Discussion
The present study revealed several major findings: (1) AL protected ISO-induced against myocardial damage in vivo; (2) AL effectively reduced CoCl2-induced apoptosis, oxidative stress, mitochondrial damage, and calcium overload in vitro; and (3) the cardioprotective effects of AL via the direct inhibition of ICa-L in rat ventricular myocytes. To our knowledge, this is the first study to investigate the mechanism of AL to protect the heart against ischemic injury.
AL has been used as one of the main components of Guanxinsuhe capsules (GXSHC). GXSHC can protect H2O2 injured cardiomyocytes by increasing their antioxidant capability. 30 Meanwhile, GXSHC treats myocardial infarction by regulating troponin. 31 Taken together, these studies indicate that GXSHC has a protective effect on the heart. However, little is known regarding whether AL can protect against myocardial disease. The findings described in the present study support the cardioprotective effect of AL against MI injury.
The pathophysiological mechanisms of MI are complex and have a profound impact on the global burden of heart disease. 32 Unlike in other tissues and muscles subtypes, cardiac metabolism is almost entirely aerobic. Therefore, the perfect balance between myocardial oxygen consumption and myocardial oxygen supply is necessary for maintaining ventricular function and avoiding MI. 33 ISO is often used to model MI in experimental animals because it induces ischemia, hypoxia, Ca2+ overload, and depletes energy reserves. 34
Examination of the ECG, LV function, and histology in rats treated with ISO showed myocardial cell damage, depressed systolic pressure, and increased LVEDP, leading to MI. Compared with the CON group, subcutaneous injection of ISO-induced MI as indicated by tachycardia and J-point elevation. Pretreatment with AL could decrease heart rate and J-point. Compared with the parameters in the ISO group, LVEDP decreased while LVSP and ± dp/dtmax significantly increased in the H-AL and L-AL groups. The evaluation of CK and LDH levels has been the gold standard for the enzymatic diagnosis of acute MI. cTnI is a polypeptide involved in myocardial contraction and is a highly sensitive biomarker of myocardial injury. In the ISO group, the levels of CK, LDH, and cTnI remarkably increased compared with the CON group. Pretreatment with AL caused the levels of CK, LDH, and cTnI to decrease. 35 Meanwhile, HE-stained sections confirmed that the heart tissue of the model group was markedly injured. Tissue from the ISO group exhibited infiltrating inflammatory cells, widened muscle space, and striation loss with nuclear changes. The damage observed in the H-AL and L-AL groups was reduced compared with the ISO group. In the present study, the mechanism underlying the myocardial protection conferred by AL was investigated by analyzing the expression levels of pro-inflammatory markers. ISO induced a significant increase in IL-6 and TNF-α expression. However, AL treatment decreased these elevated pro-inflammatory factor expression levels. Taken together, these data describe the ability of AL pretreatment to prevent the changes associated with this specific disease state.
CoCl2-treated H9c2 cells represent a robust in vitro model for exploring the mechanisms of H/I-related damage, since H9c2 cells exhibit the biochemical and electrophysiological characteristics of cardiomyocytes. 36 The H9c2 cell line has been established from embryonic rat cardiac ventricle and it has properties similar to neonatal and adult cardiomyocytes, which can functionally express LTCCs of cardiac. 37 In the current study, CoCl2 was used to mimic H/I-induced apoptosis in H9c2 cardiomyocytes. Myocardial cell apoptosis is an important pathological process in H/I-induced myocardial injury, which is closely related to cardiac insufficiency. According to the CCK-8 results, cardiomyocyte viability decreased following treatment with CoCl2, and the apoptosis rate increased. In apoptotic cells, intracellular LDH and CK are released through damaged cell membrane structures. Therefore, it is an indirect method to evaluate H9c2 cell injury by detection of LDH and CK concentration. Levels of the indicators used for MI diagnosis, CK and LDH were significantly increased after treatment with CoCl2. Importantly, we found that 5 and 10 μmol/L AL protected H9c2 cells against CoCl2-induced injury, enhancing cell viability and decreasing the levels of LDH and CK. In addition, the flow cytometry and apoptotic fluorescence results show that the AL treated groups exhibited decreased apoptosis rates, which supports the protective effect of AL against CoCl2-induced apoptosis.
Increased production of ROS and ROS-mediated oxidative damage during the MI process play crucial roles in the development of H/I injury. When ROS accumulate continuously and the endogenous antioxidant defense system cannot clear these in a timely fashion, oxidative stress is induced. 38 The cytotoxicity induced by CoCl2 is due to the increased production of free radicals (including ROS) mediated by hypoxia. The balance between the production of ROS and the elimination of excess ROS is essential to maintain the redox state and homeostasis in cells. Compared with the control cells, the CoCl2 treated cells exhibited significantly increased production of ROS. Our results showed that pretreatment with 5 and 10 μmol/L AL provided a free radical scavenging property. Mammals eliminate ROS using reducing enzymes such as SOD, CAT, and GSH; therefore, concentrations of these “free-radical scavengers” reflect cells’ ability to neutralize free radicals and reduce oxidative stress. SOD is one of the primary enzymes used by cells to maintain oxidative homeostasis. 39 GSH is also a major cellular antioxidant that serves not only as a direct electron donor to neutralize H2O2 and lipid peroxide, but also the scavenger of oxygen and nitrogen radicals. 40 CAT protects against toxic oxygen metabolites. 41 Under standard conditions, H9c2 cardiomyocytes contain high levels of SOD, CAT, and GSH. However, when cells are exposed to CoCl2-induced H/I injury, these antioxidant defense systems are destroyed, and both the level and concentration of SOD, CAT, and GSH might decrease. Additionally, MDA is the end product of polyunsaturated fatty acids and is often used to evaluate cardiac lipid peroxidation. 42 In the present study, the CoCl2 group had increased MDA content and decreased levels of CAT, SOD, and GSH. However, pretreatment with AL significantly reduced the content of MDA and restored SOD, CAT, and GSH activity. These results suggested that the cardioprotective effect of AL might be attributable to its antioxidative activity.
Myocardial mitochondria are vulnerable to damage induced by ischemia, which can lead to cardiac dysfunction. 43 In the heart, a shift in mitochondrial permeability can result from the mitochondrial dysfunction caused by oxidative stress or other stimuli, a disruption of the MMP. 44 Oxidative stress and the generation of ROS can lead to abnormal mitochondrial function. 45 In this study, the exposure of H9c2 cardiomyocytes to hypoxia led to mitochondrial damage that significantly improved with AL pretreatment.
Excessive ROS disrupt the structure of the cell membrane, increase the permeability of cell membrane, and lead to large amounts of calcium influx from the extracellular environment, resulting in intracellular calcium overload. 46 This ROS-induced increase in Ca2+ is a constant feature of pathological states related to oxidative stress. 47 Ca2+ released from the sarcoplasmic reticulum through the ryanodine receptor (RyR) also contributes to elevated intracellular Ca2+ concentrations. The excessive increase of Ca2+ concentration in cells can lead to calcium overload and therefore result in damage to cardiac cells. 48 Preventing the rise in intracellular Ca2+ concentration could reduce cardiac damage. 49 To detect the accumulation of Ca2+ in cells after hypoxia/ischemia-induced, the fluorescence intensity of Ca2+ marked by Fluo-3/AM was observed under a confocal microscope. As shown in Figure 6, exposure of H9c2 cells to H/I conditions resulted in a significant increase in intracellular Ca2+ fluorescence indicated by Fluo-3/AM. Pretreatment with AL attenuated this fluorescence intensity. Our results suggest that AL can alleviate calcium overload in H9c2 cells.
Since Ca2+ cannot be degraded, Ca2+ levels can only be controlled by transporting the ions across membranes. There are three potential sources of Ca2+ entry: (1) LTCCs. 50 (2) sarcoplasmic Na+/Ca2+ exchanger. 51 (3) another calcium entry pathway. 52 Calcium influx is predominantly carried out by LTCCs in cardiac cells. LTCCs play a central role in calcium overload, and the regulation of LTCC activity is the focus of considerable research efforts. 53 The inhibition of LTCCs leads to a decrease in Ca2+ entry, which in turn reduces harmful calcium overload, thereby protecting the heart against ischemic injury. Calcium ion blockers are the most common and widely used drugs for the treatment of MI. Therefore, the effects of AL on LTCCs in isolated rat ventricular myocytes were investigated using the whole-cell patch-clamp technique. In these experiments, VER was used as the positive control to block the LTCCs. 54 Our data suggest that AL reduces ICa-L in a concentration-dependent manner with an IC50 of 17.29 μmol/L in cardiomyocytes. AL also reduced ICa-L at 10 μmol/L in both healthy and ischemic myocardial cells. Figure 10 shows that neither the reversal potential nor the I-V relationship of ICa-L changed. Furthermore, at 3 and 10 μmol/L, AL shifted the steady-state activation and inactivation curves of ICa-L to the left. These results demonstrate that MI suppressed the ICa-L primarily by decreasing the Ca2+ current amplitude, a component of the mechanism underlying AL’s effects against MI injury.
One of the limitations of this study is that there is a gap between this experimental environment and the physiological environment of cardiomyocytes. Furthermore, in this study, AL inhibits Ca2+ inflow by suppressing gated voltage LTCCs, thus alleviate intracellular calcium overload and oxidative stress. However, cardiac LTCCs are regulated by a variety of neurotransmitters, hormones, and cytokines. Sperelakis and Schneider demonstrated that β-adrenergic receptor-mediated stimulation of cardiac LTCCs was due to phosphorylation of the channel by cAMP-dependent protein kinase A (PKA). 55 In addition, other signaling pathways have also been suggested to regulate the channel by phosphorylation. α-Adrenergic agonists, endothelin, and angiotensin II all regulate LTCCs through the protein kinase C (PKC) pathway.56,57 Thus, additional studies are needed to identify the pathways underlying AL’s impact in MI. Furthermore, whether AL acts directly on ISO (e.g. by degrading it) must be addressed in future studies on the relationship between the AL and ISO.
Conclusions
In a nutshell, the overall findings exemplify that AL alleviates cardiac injury in vivo and in vitro. The cardioprotective effects of AL may be related to decreased heart rate, ST interval, and alleviated myocardial pathological injury. Meanwhile, as shown in Figure 11, AL protects H9c2 cells against CoCl2-induced H/I injury by improving cell viability, apoptosis, LDH, and CK leakage, antioxidant defenses, mitochondrial damage, and calcium overload. In addition, our findings support that, as an LTCC blocker, AL may play a positive role in the protection of ischemic cardiomyopathy. Cardioprotective effects of AL on MI injury.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was sponsored by grants from the Foundation of Medical Science Research of Hebei Province (No. 20180886).
Ethical approval
Ethical approval for this study was obtained from the Ethics Committee for Animal Experiments of Hebei University of Chinese Medicine (approval number: DWLL2020073).
Informed consent
There are no human subjects in this article and informed consent is not applicable.
Animal welfare
The present study followed international, national, and/or institutional guidelines for humane animal treatment and complied with relevant legislation.
Availability of data and materials
The datasets used or analyzed during the current study are available from the corresponding author on reasonable request.
Statement of human and animals rights
All experimental procedures with animal subjects in this study were conducted in accordance with the Guidelines of Animal Experiments from the Committee of Medical Ethics.