
Abstract
Common variable immunodeficiency (CVID) and immunoglobulin A deficiency (IgAD) are the most prevalent primary immunodeficiency disorders. High rates of familial inheritance have been described in CVID and IgAD, but it is unknown in different ethnic populations. We aimed to determine the prevalence of familial cases and whether they showed more severe clinical characteristics than sporadic ones in Turkish patients. A total of 40 CVID and 70 IgAD patients and their 251 first-degree relatives (FDRs) were evaluated. Demographic, clinical, and laboratory data were reviewed. A familial case was defined as a patient with at least one affected FDR (A-FDR). The rate of parental consanguinity was 19.1%. There were 37 familial cases (37/110) (33.6%) with at least one A-FDR. There were 48 A-FDRs who had immunoglobulins lower than age-related normals (48/251) (19.1%). Pulmonary infections were significantly higher in familial cases. To our knowledge, this study includes the highest number of CVID/IgAD patients and their FDRs in literature. Familial cases are at least 30% of the IgAD and CVID patients, and they have more frequent lower respiratory tract infections than sporadic ones, so these patients have to be evaluated depending on their being familial or sporadic for better management. The risk of carrying any immunologic alterations in relatives of patients with IgAD and CVID is approximately 20%. Although most A-FDRs are asymptomatic, considering the risk of progression to CVID by age, we highly recommend routine screening for FDRs.
Introduction
Common variable immunodeficiency (CVID) and immunoglobulin A deficiency (IgAD) are the most common forms of primary antibody disorders in humans, with an estimated incidence of 1:25000 and 1:600, respectively.1,2 Affected individuals typically present with recurrent bacterial infections, predominantly of the respiratory tract.2,3 Patients may also suffer from gastrointestinal, autoimmune, and inflammatory disorders and malignancy.1,4
Most IgAD and CVID cases are sporadic, but some cases have at least one additional member suffering from CVID or IgAD. 2 Most multiplex families show an autosomal dominant mode of inheritance but about 20% of cases present with the recessive trait. A common genetic basis for IgAD and CVID is suggested by their occurrence in members of the same family and similarity of the underlying B-cell differentiation defects. In addition, some affected individuals initially present with IgAD and then show progression to CVID.1,3,4 A disease-causing gene for an autosomal dominant CVID/IgAD has been found on chromosome 4q. 5 Rare autosomal mutations in single genes, namely TACI, ICOS, BAFF-R, CD19, CD20, CD81, and MSH5, have been reported in CVID.5,6 The precise molecular cause for IgAD is also still unknown. Numerous studies of these complex multifactorial disorders have shown human leukocyte antigen (HLA) associations, in particular, with the HLA haplotypes B8, DR3, DR7, and DQ2.5,6
In this study, we aimed to determine the prevalence of familial IgAD and CVID cases and whether familial cases (FCs) show more severe clinical characteristics than sporadic ones. Second, we screened first-degree relatives (FDRs) and determined the prevalence and clinical findings of affected FDRs (A-FDRs).
Patients and methods
The study population consisted of CVID (n = 40) and IgAD patients (n = 70) who fulfilled criteria for CVID and IgAD from the out-patient and in-patient clinics of Ege University Faculty of Medicine, Department of Pediatric Immunology, Turkey, and their FDRs (n = 251). Patients were diagnosed and classified according to the European Society for Immunodeficiencies/Pan-American Group for Immunodeficiency (ESID/PAGID) criteria. 7
Diagnostic criteria for CVID were as follows: (1) marked decrease of IgG (at least 2 standard deviations (SDs) below the mean for age, measured at least twice), (2) reduced serum IgA and/or IgM, (3) absent isohemagglutinins and/or impaired response to vaccination, (4) onset of immunodeficiency at greater than 2 years of age, and (5) exclusion of other known causes of hypogammaglobulinemia.
IgAD patients were also divided into two groups as selective and partial IgAD. Selective IgAD was defined as a blood IgA below 7 mg/dL with normal IgG and IgM levels in patients over 4 years of age. Partial IgAD is defined as IgA levels which is lower than mean − 2SD of age-related normals.
All demographic information including name, gender, date of birth, age at onset of symptoms, age on admission, age at diagnosis, family history and consanguinity, clinical symptoms or complications (autoimmune disease, chronic giardiasis, granulomatosis, allergy, lymphoma or any other malignancy, lymphadenomegaly, splenomegaly, bronchiectasis, musculoskeletal system findings, celiac-like disease), follow-up duration, and laboratory data were recorded from medical files. Parents provided written informed consent before the study.
An FC was defined as a patient with at least one A-FDR. FDRs with low immunoglobulin levels and relatives with normal immunoglobulin levels were classified as A-FDR (affected FDR) and H-FDR (healthy FDR), respectively. Patients who did not show any decreased immunoglobulin levels in family screening were accepted as sporadic cases (SCs). A questionnaire including demographic data (age, gender, consanguinity) and clinical signs was administered to all relatives.
Serum immunoglobulin levels (IgG, IgA, IgM) were analyzed quantitatively by Dade Behring BNII nephelometer, Siemens, Germany, and compared with normal levels obtained from almost 500 healthy children in 14 different age groups from the same ethnic origin (data were published previously 8 ).
All clinical and laboratory data were evaluated in relation to each other and compared in different patient profiles and control groups. Statistical analyses were performed using SPSS Windows Version 17.0, SPSS Inc., Chicago, IL, USA. Data were expressed as mean plus or minus SD except where indicated otherwise. Correlation comparisons between paired samples were made by Pearson’s correlation coefficient. Statistical comparisons of numeric data were made using Student’s t test and classified data were evaluated by chi-square test. A two-sided P < 0.05 showed statistical significance.
Results
The study included 361 participants (110 cases, 251 FDRs). Demographic, clinical, and some laboratory features of IgAD (n = 70) and CVID (n = 40) patients are shown in Table 1. Although age at the beginning of the symptoms did not differ, the mean age of the study group and age of CVID patients at diagnosis were higher than IgAD patients, P = 0.000 and P = 0.009, respectively. The rates of parental consanguinity (P = 0.000) and a positive family history of a known immunodeficiency (P = 0.003) were statistically significant between groups; they were more prevalent in CVID patients (Table 1). Positive family history was not present in the selective IgAD group.
Demographic, clinical, and some laboratory findings of patients.
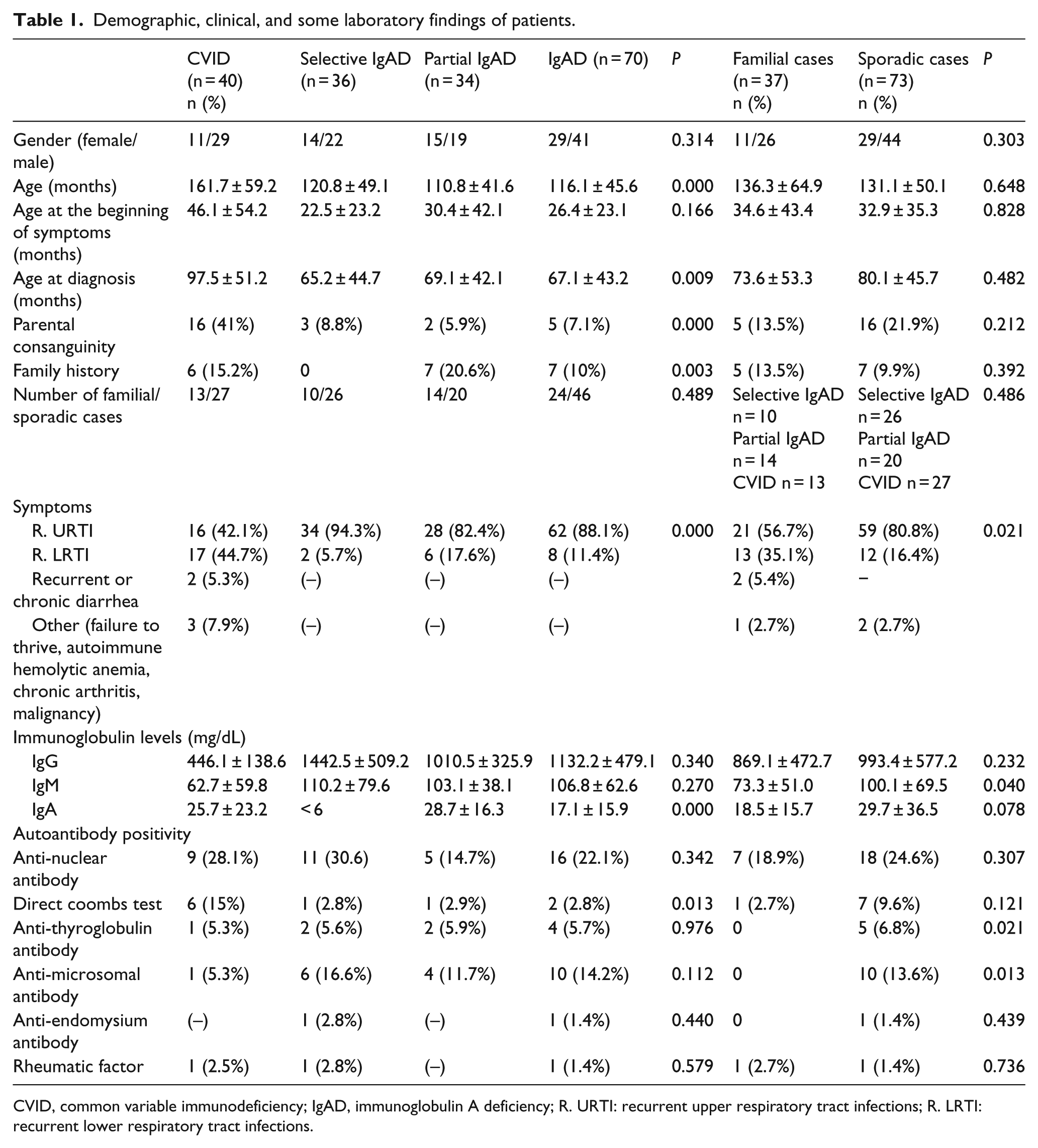
CVID, common variable immunodeficiency; IgAD, immunoglobulin A deficiency; R. URTI: recurrent upper respiratory tract infections; R. LRTI: recurrent lower respiratory tract infections.
Recurrent respiratory tract infections were the leading cause of hospital admissions in all groups. The frequency of lower respiratory tract infections, chronic or recurrent diarrhea, and other clinical signs such as failure to thrive, autoimmune hemolytic anemia, and chronic arthritis were higher in CVID group. Complications observed in CVID patients during follow-up were as follows: bronchiectasis (20%), splenomegaly (25%), osteoporosis (30%), delayed growth (30%), autoimmunity (autoimmune hemolytic anemia, immune thrombocytopenia, sacroiliitis, vasculitis) (12,5%), hepatomegaly (25%), gluten-like enteropathy (10%), granuloma formation (15%), and malignancy, namely acute myeloid leukemia and non-Hodgkin lymphoma (5%). None of the IgAD patients experienced clinically overt autoimmune or inflammatory complications. The anti-nuclear antibody, anti-thyroglobulin antibody, anti-microsomal antibody, anti-endomysium antibody, and rheumatic factor positivities between disease groups were statistically insignificant (Table 1). Fifteen percent of CVID patients (n = 6) had positive direct Coombs test, two out of these had autoimmune hemolytic anemia.
IgG levels of selective IgAD patients were very high and the mean levels of all the three immunoglobulins were low in CVID patients. The mean IgG, IgA, and IgM levels of FDRs were almost similar and normal, although we found several low levels in individual investigations. There were 48 FDRs (A-FDR) who had immunoglobulins lower than mean − 2SD of age-related normals (48/251) (19.1%) (Table 2).
Distribution of familial and sporadic cases, affected and healthy family members in our study group compared to other populations in previous studies.

IgAD, immunoglobulin A deficiency; CVID, common variable immunodeficiency; FC, familial case; SC, sporadic case; FDR, first-degree relative; A-FDR, affected FDR; ND, not determined.
Total counts of FC, SC, A-FDR, and H-FDR cases concerning the diagnostic groups are shown in Table 2. There were 37 FCs (37/110) (33.6%) who were found to have at least one A-FDR. The ratios of FCs were 27.7% (10/36), 41.1% (14/34), and 32.5% (13/40) in selective IgAD, partial IgA, and CVID patients, respectively. FC cases were 34.2% in total IgAD, and 32.5% in CVID patients and these ratios did not show any significant difference as well as in SC cases (P = 0.489) (Table 2). The ratio of A-FDRs was exactly same in IgAD and CVID groups (19.1%) as well as in selective (19.0%) and partial IgAD (19.2%) cases (Table 3).
Immunologic abnormalities detected in relative-FDRs of patients with selective IgAD, partial IgAD, and CVID.

A-FDR, affected FDR; FDR, first-degree relative; IgAD, immunoglobulin A deficiency; CVID, common variable immunodeficiency.
The frequencies of diagnosis, gender, and parental consanguinity among FCs and SCs were statistically insignificant (Table 1). Pulmonary infections were significantly higher in FC (35.1%) than SC (16.4%). There was no statistically significant difference in IgG and IgA levels between FCs and SCs (Table 1). IgM was significantly decreased in FCs (P = 0.040, Table 1). There is a significant correlation between the patients with the rate of pulmonary infections and low IgM levels (r = –0.203, P = 0.036). Higher prevalence of positive anti-thyroglobulin and anti-microsomal antibody titers were observed in SCs, with no evident clinical or radiological manifestations of autoimmune thyroiditis.
Forty-eight (19.1%) cases of FDRs showed immunologic abnormalities (Table 3), 39.5% (19) were mothers, 37.5% (18) were fathers, and 22.9% (11) were siblings. The main abnormality was IgAD (selective IgAD n = 28, partial IgAD n = 10) in A-FDRs (Table 3). Eight cases of FDRs had IgM deficiency. IgG deficiency (n = 2) among FDRs was rare. One mother (IgG = 612 mg/dL) and one father (IgG = 488 mg/dL) in CVID group had asymptomatic hypogammaglobulinemia in adulthood. Both of them had recurrent upper respiratory tract infections during school ages, which improved spontaneously by age. One selective IgAD mother of a CVID case had Hashimoto’s thyroiditis. One father (with decreased IgM level) of another CVID patient had vitiligo and a history of recurrent sinusitis. All patients with affected siblings also had parental (paternal or maternal) IgAD. Gastrointestinal symptoms, hospital admissions, and chronic treatment for different diseases were significantly increased in A-FDRs when compared with H-FDR group.
Discussion
Parental consanguinity and the autosomal recessive mode of inheritance are risk factors for primary immunodeficiency disorders.9,12,13 In Rivoisy et al.’s 14 study, CVID patients who had consanguineous parents had more severe complications like splenomegaly, granulomatous disease, and bronchiectasis. In our study, parental consanguinity and positive family history rates were 19.1% and 11.8%, respectively. These rates were significantly higher in CVID patients than IgAD patients. Positive family history of IgAD and CVID was pointed out as the most significant risk factor for developing the disease by Vorechovský et al. 15 An Iranian CVID study showed that 20% of relatives had hypogammaglobulinemia. 9 In Rezaei et al.’s 10 study, 11.3% of relatives of CVID and IgAD patients showed abnormal immunoglobulin levels in forms of CVID, sIgAD, or IgG2 deficiency. The risk of being a patient was reported to be significantly higher among siblings than parents.9,10,15 Soler-Palacín et al. 11 screened 88 FDRs of 42 sIgAD patients. The risk of IgAD in FDRs was slightly higher in mothers. In our study, there were a total of 37 FCs and 48 A-FDRs. The rate of affected mothers was the highest and found as 19.7%. Four different studies9–11 were compared in Table 2. The rate of FCs is almost the same whereas approximately 30% of patients with CVID and/or IgAD are FCs suggesting that at least 30% cases may have a genetic basis and a shared molecular defect.
Azizi et al. 3 reported that the most prevalent presentations of immunodeficiency were respiratory tract infections and chronic diarrhea in primary antibody deficiency patients. In our study, the frequency of lower respiratory tract infections, chronic diarrhea, and complications such as bronchiectasis, autoimmunity, and failure to thrive were more common in CVID group than IgAD group. Frequent respiratory tract infections in FCs could be related to lower IgM levels.
There was not any significant difference in the ratio of affected family members with respect to disease groups, namely CVID, and selective and partial IgAD groups. A total of 48 FDRs (19%) were found to have an immunologic abnormality. In Aghamohammadi et al.’s 9 study, IgG levels in all family members of CVID patients were normal and the most common alteration in humoral immunity was IgM deficiency. In our study, most of the A-FDRs were asymptomatic, and the most common humoral abnormality was IgAD. Symptomatic A-FDRs had recurrent upper respiratory tract infections, sinusitis, vitiligo, and Hashimoto’s thyroiditis.
In conclusion; at least 30% of the IgAD, and CVID patients are familial and they have more frequent lower respiratory tract infections than sporadic ones, so that these patients have to be further evaluated with respect to their being familial or sporadic for better management. The probability of immunologic alterations in relatives of these patients is approximately 20%. Although most of them are asymptomatic, considering the increased risk gradual progression to CVID, we highly recommend routine screening and tailored approach to the FDRs of IgAD and CVID patients.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.