
Abstract
Background
Neurofibromatosis type 1 (NF1) is an autosomal dominant genetic neurocutaneous disorder. Multiple sclerosis (MS) is an immune-mediated demyelinating disorder of the central nervous system. The rates of occurrence are higher than expected by chance, suggesting that an underlying predisposition may exist. We aimed to systematically review the literature to better understand the concurrence and contribute four additional patients to the existing literature.
Methods
This study is a systematic review as well as retrospective case series. Pubmed, Embase, Cochrane, Scopus, and Web of Science were queried for full articles in English reporting on cases of concurrent NF1 and MS between 1990 and 2023.
Results
The systematic review revealed 34 reported cases of patients with both NF1 and MS across 18 papers. Ten patients had primary progressive MS, seven had relapse-remitting MS, five had secondary progressive MS, and 12 did not have a specific MS classification. Thirty-three had at least one reported brain lesion and 18 had at least one reported spine lesion. Our cohort contains four female patients; all with brain lesions and three with spinal lesions.
Conclusion
Although rare, NF1 and MS can occur within the same patient. Previous literature has been inconsistent in the information reported about patients with these concurrent diagnoses. Additional research is required to better understand the concurrent disease processes, whether a predisposition exists, as well as the best therapeutic approaches for these patients.
Introduction
Neurofibromatosis type 1 (NF1) results from heterozygous germline alterations in NF1, a tumor suppressor gene which encodes the GTPase-activating protein neurofibromin, with subsequent somatic loss of function mutations causing tumors. NF1 is the most common neurocutaneous disorder, with a pooled prevalence estimated at one in 3190 worldwide. 1 Available data suggest that NF1 is observed with similar frequency across ethnic groups. 2 NF1 is an autosomal dominant genetic disorder, though approximately half of new cases of NF1 result from de novo mutations. 3
NF1 is completely penetrant but phenotypically heterogeneous, with various systemic as well as neurologic manifestations. People with NF1 are at increased risk for tumor development, including the hallmark of NF1: neurofibromas. NF1 is diagnosed clinically, based on the presence two or more features including: six or more café-au-lait macules (CAL), axilla or inguinal skinfold freckling, two or more iris Lisch nodules, two or more choroidal abnormalities seen on optical coherence tomography or near-infrared reflectance imaging, characteristic bone lesions, optic pathway glioma, two or more neurofibromas of the skin or one plexiform neurofibroma, a first degree relative with NF1, and a pathogenic variant of the NF1 gene. 4
Multiple sclerosis (MS) is the most common inflammatory demyelinating disorder of the central nervous system. Though the exact cause of MS is not fully understood, it is thought to be immune-mediated and resultant from an interplay of genetic and environmental factors. The overall prevalence of MS is estimated at one in 323 adults in the United States, with a preponderance in latitudes of increased distance from the equator. 5 Prevalence is higher in women, peaks in the age range of 35–64, and declines in those 65 years of age or greater.5,6 A large retrospective cohort study in southern California demonstrated that age- and sex-standardized prevalence is equally high in people who are African American/Black as people who are White, and lower in people who are Hispanic or Asian. 6 Low vitamin D levels, cigarette smoking, and history of Epstein-Barr viral infection have also been implicated in increased risk of MS, however these have yet to be fully elucidated in mechanism and have not been proven causal.7–9 Though there have been over 200 polymorphisms associated with additive increased risk of MS, no single variant has been found to be necessary nor sufficient. 10
Clinically MS is characterized by temporally dispersed multifocal inflammatory lesions of the brain and/or spinal cord. Active lesions cause demyelination and often episodes of acute or subacute focal neurologic symptoms. Highly characteristic acute or subacute clinical manifestations include optic neuritis, focal motor weakness or sensory changes, bladder dysfunction, gait instability, or a brainstem syndrome among others. Such “attacks” of acute symptoms often remit over time, though not always or completely. Chronic lesions are often characterized by axonal degeneration. Chronic symptomatic manifestations include pain, fatigue, and progressive neurodegeneration. MS is diagnosed based on the revised McDonald criteria, which requires evidence of lesion dissemination in space and time, using clinical history accompanied by imaging findings and/or presence of oligoclonal bands on cerebral spinal fluid analysis. 11 Typical magnetic resonance imaging (MRI) findings of MS include T2 hyperintense white matter lesions in characteristic locations (including periventricular, cortical, juxtacortical, infratentorial, or spinal cord regions). Active lesions are often T1 contrast-enhancing with gadolinium.
NF1 has been posited to have a link with several neurological disorders, including MS. 12 There has been documentation of co-occurrence of NF1 and MS in the literature. 13 One retrospective study looking at administrative healthcare claims found higher odds of healthcare claims related to MS in those patients with NF1 than those without. The study looked at 8579 people with NF1 and 85,790 without NF1 based on a large database of deidentified insurance data claims. MS occurrence in the NF1 group was higher; 25 (0.3%) versus non-NF1 group 108 (0.1%), OR 1.9, CI [1.2–2.9]. 14 Additionally, incidences of co-occurrence of NF1 and MS have been previously reported in the literature in case reports or small case series. 13
There is some theoretical basis to the idea that NF1 and MS may co-occur more frequently by chance alone. NF1 is a tumor suppressor gene that encodes neurofibromin, which regulates the Ras cellular proliferation pathway via GTPase.15,16 Located on chromosome 17.11.2q, NF1 is one of the largest genes in the human genome, with 55 constitutive exons and five alternatively spliced exons spanning about 350 kb. 17 Neurofibromin is expressed in high abundance in neurons as well as oligodendrocytes and Schwann cells. 18 Myelin sheaths formed around the axons of neurons are essential to proper and rapid conduction of action potentials in the nervous system. Schwann cells are integral to the process of myelination in the peripheral nervous system, and oligodendrocytes are responsible for the formation and maintenance of myelin in the central nervous system. Several membrane glycoproteins are integral to this process, oligodendrocyte-myelin glycoprotein (OMgp) among them.19,20 The OMgp gene lies within intron 27b of NF1, with its protein transcribed in the opposite direction as neurofibromin. 21 Demyelination in the central nervous system is a hallmark of MS, as is loss of oligodendrocytes. There is some reason to consider then, that either an impairment in OMgp or loss of high expression of neurofibromin in oligodendrocytes could predispose individuals with NF1 to MS.
Given the large numbers needed to evaluate such a relationship, currently there is a paucity of information regarding the clinical co-occurrence of NF1 and MS. This article provides a systematic review of the existing case literature as well contributing four novel patients from our institutional cohort.
Methods
This study is a systematic review of previously published cases and case series as well as a retrospective case series.
For the systematic review the investigators (D.B. and C.R.) created an initial search strategy that was then refined by a medical librarian (M.S.), to search for full articles in English reporting on cases of concurrent NF1 and MS between 1990 and 2023. The Covidence platform was used for compilation of the systematic review. Pubmed, EMBASE, Cochrane, Scopus, and Web of Science were queried. The search was conducted on 10 November 2023. Search terms included neurofibromatosis 1, NF1, multiple sclerosis, MS, neuromyelitis optica, MOG, and MOGAD. The authors (D.B. and G.T) then independently screened all titles and abstracts identified by the search. Full-text versions were then obtained for the remaining article and assessed for relevance.
For the case series, IRB waiver of HIPAA privacy authorization was obtained from the Johns Hopkins University School of Medicine (IRB 00403582). Additionally, all participants provided verbal consent to be included in this study.
Study selection
Studies reporting cases or series of cases with patients diagnosed with both NF1 and MS were included. Review articles that also contributed novel case(s) were included. Cohort studies and prospective clinical trials were not included. No meta-analyses were included. Articles that referenced cases reported in one of our other included articles were not excluded, but the patient cases referenced were not added to data extraction to avoid duplication. Only English-language articles and articles with full text available were considered.
Data extraction
The authors (D.B. and G.T) performed independent data extraction on the finalized list of studies. The information extracted included: (where available) sex, age at diagnosis, MS subtype, presence of MS brain lesions or spine lesions, disease modifying therapy (DMT).
Articles that referenced cases reported in one of our other included articles were not excluded, but the patient cases referenced were not added to data extraction to avoid duplication.
In cases of disagreement the authors (D.B. and G.T) discussed and came to consensus.
Results
Systematic review
One hundred ninety unique studies were returned from the search. One hundred sixty-three of these were excluded in initial abstract and title review. Full-text versions were obtained for the remaining 27 studies, which were then assessed for eligibility with a full-text review. Full-text review resulted in the exclusion of nine additional studies. Eighteen studies ultimately met criteria for inclusion of this work (Figure 1). Data were subsequently extracted by hand (summarized in Table 1).

Preferred reporting items for systematic reviews and meta-analyses (PRISMA) flow diagram depicting the flow of information through the different phases of the systematic review.
Systematic review data extraction of the preexisting case studies of individuals with NF1 and MS.
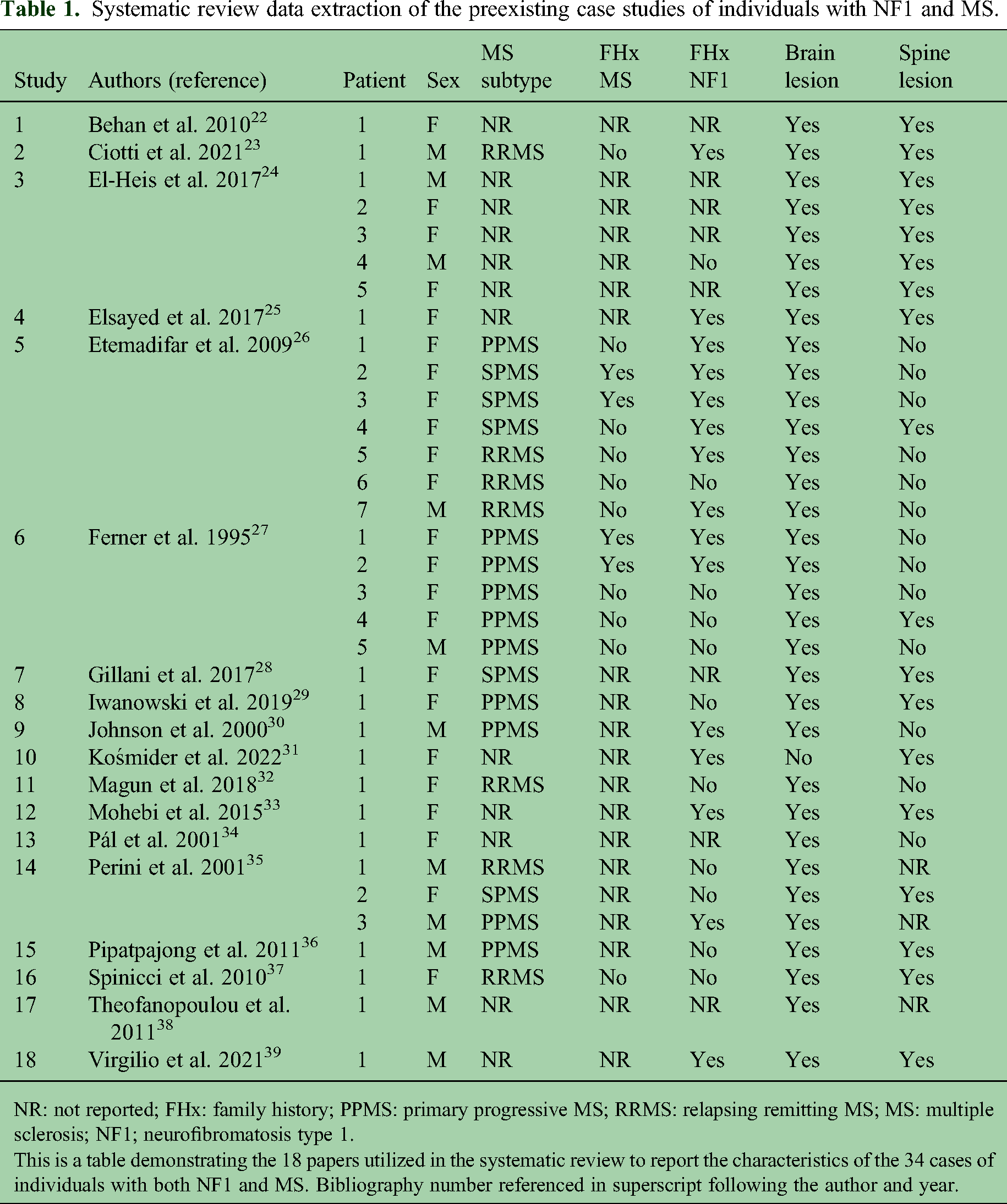
NR: not reported; FHx: family history; PPMS: primary progressive MS; RRMS: relapsing remitting MS; MS: multiple sclerosis; NF1; neurofibromatosis type 1.
This is a table demonstrating the 18 papers utilized in the systematic review to report the characteristics of the 34 cases of individuals with both NF1 and MS. Bibliography number referenced in superscript following the author and year.
There were 34 reported cases of patients with both NF1 and MS in the literature across 18 papers. Twenty-three patients were female and 11 were male. Thirty-two of 34 patients were first diagnosed with NF1. Only one patient had a preexisting diagnosis of MS when diagnosed with NF1, and one patient was diagnosed with NF1 and MS simultaneously. Ten patients had primary progressive MS, seven had relapse-remitting MS, five had secondary progressive MS, and 12 did not have a specific MS classification. Thirteen had a family history of NF1, though six did not have data about NF1 family history. Thirty-three had at least one reported MS-related brain lesion and 18 had at least one reported MS-related spine lesion. Ten patients received DMT for MS. Three received interferon beta-1a. One received each of the following: interferon Beta-1a and azathioprine, azathioprine, dimethyl fumarate, natalizumab, mitoxantrone, methotrexate. One received an unreported therapy. Age (for either time of assessment or time of diagnoses) was not uniformly reported.
Case series
Four patients were included, data is summarized in Table 2.
The characterization of NF1 and MS in our single institutional cohort.

NR: not reported; DMT: disease modifying therapy; KPS: Karnofsky Performance Scale; IEP: independent education plan; PPMS: primary progressive MS: RRMS: relapsing remitting MS.
This table characterizes the demographic, diagnostic criteria met for NF1, diagnostic criteria met for MS, treatment of MS, and KPS for our institutional cohort of four patients.
Patient 1
Forty-one-year-old woman diagnosed with NF1 at age 4 based on cutaneous manifestations and presence of a plexiform neurofibroma, including the presence of >6 CAL macules, skin fold freckling, cutaneous neurofibromas, Lisch nodules, and a lower extremity plexiform neurofibroma. She has no family history of NF1. NF1 related management included a debulking surgery of the lower extremity plexiform neurofibroma in childhood. No history of radiation, chemotherapy, or targeted medication therapy.
She was diagnosed with MS at age 38 due to incidental MRI brain findings of a left parietal periventricular T2 hyperintense white matter lesion with punctate enhancement as well as additional small periventricular, pericallosal, and subcortical white matter T2 hyperintense white matter lesions meeting the criteria for dissemination in space (Figure 2). The patient was asymptomatic at the time of her diagnosis, but upon review of her neurologic history, she reported prior episodes of transient neurologic symptoms (distinct from those of her known cutaneous NF1 lesions and her left lower extremity paresthesia associated with the plexiform neurofibroma) that met the criteria for dissemination in time. She thus met the 2017 revised McDonald criteria for MS. She has never been initiated on DMT due to shared decision making, which was felt to be reasonable given the lack of new or worsening neurologic deficits or radiologic signs of progression. She has no family history of MS.
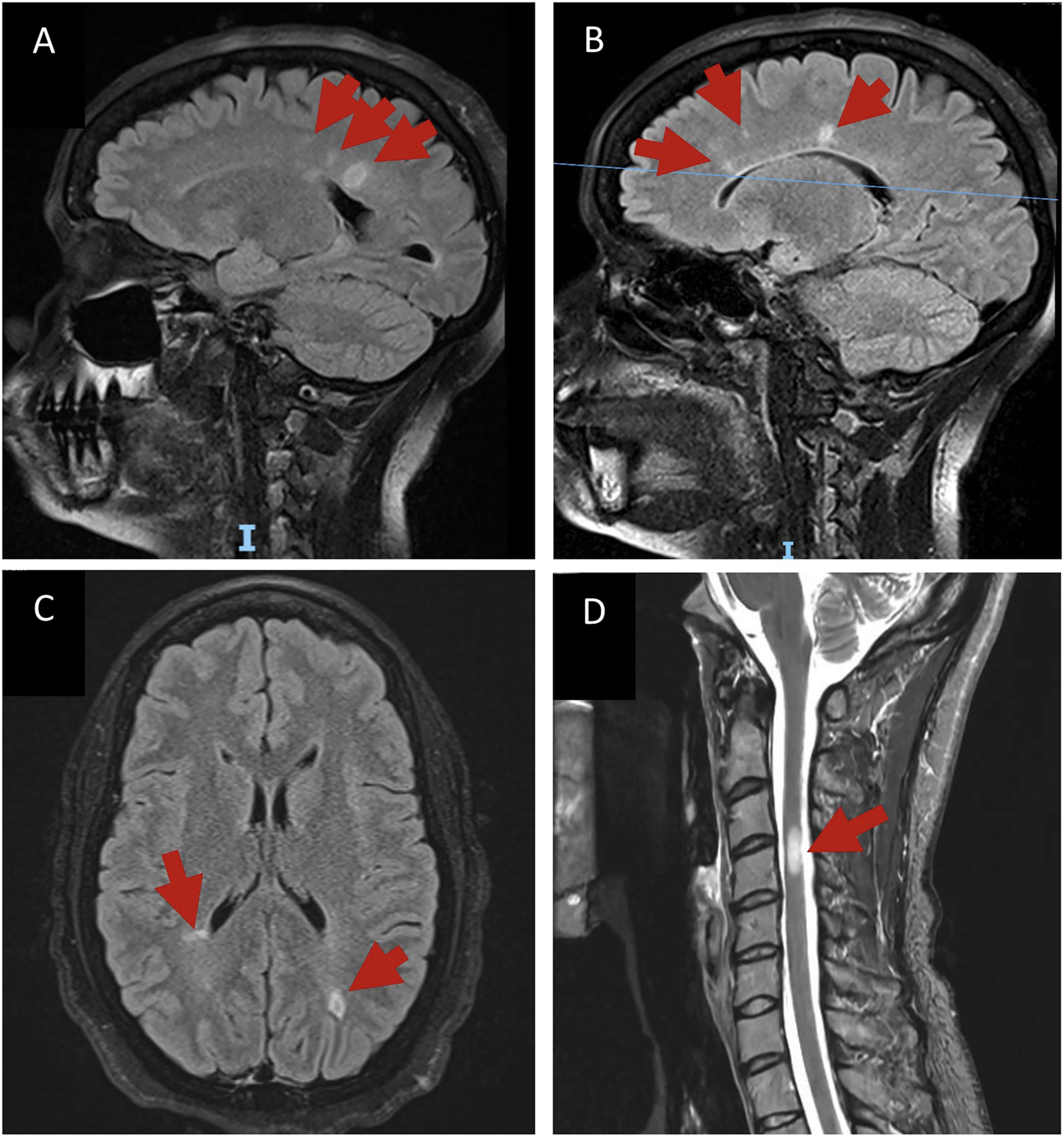
T2/FLAIR MR images. (A) Sagittal section obtained from patient number 1 demonstrating a left parietal periventricular white matter lesion adjacent to the posterior body of the lateral ventricle, and (B) radiating from the corpus callosum, typical for MS (also known as Dawson fingers). (C) Axial view of periventricular and subcortical MS plaques. (D) From patient number 2, T2 hyperintense lesion the cervical spinal cord without mass effect or perilesional edema.
She has a history of remote thyroid cancer treated with thyroidectomy, with subsequent metastatic disease to the lungs treated with radioactive iodine. She remains on active surveillance with visits to oncology every six months. No history of tobacco use. She holds a college degree and no reported history of learning problems. Self-reported race and ethnicity is Black, non-Hispanic. She is independent in all activities of daily living (ADLs). Her Karnofsky Performance Status (KPS) was 90% at her most recent clinic visit.
Patient 2
Forty-year-old woman diagnosed with NF1 as an infant based on cutaneous manifestations, including the presence of >6 CAL macules, and Lisch nodules. She later developed cutaneous neurofibromas and a left lower extremity plexiform neurofibroma. She has no family history of NF1. NF1-related management has included surgical treatment of cutaneous neurofibromas. No history of radiation, chemotherapy, or targeted medication therapy.
She was diagnosed with MS at age 40 after investigation for intermittent, transient neurologic symptoms including an episode of vertigo and an episode of extremity pruritis and spasms. She has no family history of MS. MRI brain with nonenhancing T2 hyperintense juxtacortical, periventricular, and supratentorial white matter lesions. MRI spine with nonenhancing T2 hyperintense lesions in the cervical cord as well as the thoracic cord. Appearance of lesions thought to be more consistent with demyelinated plaques than focal areas of high signal intensity (FASI) by neuroradiology and neurology teams. Lumbar puncture additionally demonstrated restricted oligoclonal bands. She met the 2017 revised McDonald criteria for MS, subclassified as relapsing-remitting MS, and was initiated on DMT with ocrelizumab. This was stopped after one dose due to intolerable fatigue and she remained off therapy for about one year. She was started on dimethyl fumarate after a new area of T2 hyperintensity was appreciated in her thoracic spinal cord MRI and imaging has been stable since that time.
Prior history of tobacco use. She holds a college degree. She has a reported history of IEP in elementary school. Self-reported race is African American and white, for ethnicity she preferred not to disclose. She is independent in all ADLs. Her KPS was 90% at her most recent clinic visit.
Patient 3
Sixty-five-year-old woman diagnosed with NF1 as an infant based on cutaneous manifestations, including the presence of >6 CAL macules, and cutaneous neurofibromas, as well as a family history of NF1 affecting her father and sibling. She later developed Lisch nodules, cutaneous and subcutaneous neurofibromas, and a plexiform neurofibroma in the left extremity as well as her back. No history of radiation, chemotherapy, or targeted medication therapy for NF1.
She was diagnosed with MS at age 56 after investigation for intermittent, transient neurologic symptoms of dizziness. Looking back, she reported decades of transient focal neurologic symptoms as well. She has no family history of MS. Records from her original diagnosis, including MRI and oligoclonal bands, were not available as these occurred at an external institution. MRI brain and spinal cord imaging available from establishment of care in our system demonstrated multiple nonenhancing T2 hyperintense juxtacortical and periventricular white matter lesions, as well as multiple nonenhancing T2 hyperintense lesions in the cervical and thoracic spine. Based on her imaging and clinical history she met the 2017 revised McDonald criteria for MS, and was subclassified as relapsing-remitting MS. She was initiated on DMT with glatiramer acetate at the time of her initial diagnosis, and was switched to Siponimod after seven years due to progressive symptoms and imaging changes by her external provider. Symptomatically, she currently describes slow progression in fatigue and imbalance overtime. This is punctuated by infrequent periods of subacute worsening and incomplete recovery consistent with relapses. She is followed in MS clinic and per report patient is considering agreeing to a switch to another DMT.
No history of tobacco use. She holds a college degree and a doctorate. She was diagnosed with attention deficit disorder in childhood. Self-reported race is White, and ethnicity is Hispanic. She is independent in all ADLs. Her KPS was 90% at her most recent clinic visit.
Patient 4
Fifty-three-year-old woman diagnosed with NF1 as an infant based on cutaneous manifestations, including the presence of >6 CAL macules and cutaneous neurofibromas, as well as a family history of NF1 affecting her father and sibling. She later developed cutaneous and subcutaneous neurofibromas, and a sacral peripheral nerve sheath tumor. NF1-related management has included a surgical removal of a facial neurofibroma. No history of radiation, chemotherapy, or targeted medication therapy for NF1.
She was diagnosed with MS at age 36 after workup for subacute gait instability and progressive bladder dysfunction. Records from her original diagnosis, including MRI and oligoclonal bands, were not available as these occurred at an external institution. MRI brain and spinal cord imaging available from establishment of care in our system demonstrated multiple periventricular nonenhancing T2 hyperintense lesions and several T1 hypointense lesions. MRI spine demonstrated cervical and thoracic cord nonenhancing T2 hyperintense lesions that are more suggestive of demyelination versus FASI as well. Based on her imaging and clinical history she reportedly met the 2017 revised McDonald criteria per her external provider, and was diagnosed with relapsing-remitting MS. She was initiated on DMT with glatiramer acetate. She has continued to have symptoms of gait instability, difficulty with balance, left lower extremity numbness and weakness, bladder dysfunction, and fatigue in a waxing and waning course. She has had multiple acute episodes of worsening fatigue, ambulation, and pain that have been treated with high dose steroids over the past decades. She also had one well-documented sudden onset intermittent episode of left foot drop, fatigue and speech changes that improved with high dose steroids. She was briefly on dalfampridine but had a provoked seizure on this medication and was subsequently discontinued (with no recurrence of seizure). She was transitioned to DMT with monomethyl fumarate at age 51, though only limited records from the external institution about her MS management and decision making were available.
No history of tobacco use. She holds a college degree. No reported learning difficulties. Self-reported race is White, and ethnicity is non-Hispanic. She is independent in all ADLs. Extrapolated from her clinical and exam documentation, her KPS was 90% at her most recent clinic visit.
Discussion
This review of the literature identified 34 unique cases of reported co-occurrence of NF1 and MS. The reports identified describe patients on multiple continents from different populations. The majority were first diagnosed with NF1 and then diagnosed with MS later in life, which is usual and in line with the known natural history and typical timeline of symptoms for both disease processes. These patients were frequently reported to be on DMT for MS. Treatment history for NF1-related complications was only sporadically reported. It is challenging to report trends in treatment practices for these co-occurring conditions, given the inconsistent reporting of data and the small sample sizes involved.
This work also contributes an additional four patients to the existing NF1 and MS literature. The cohort reported here is comprised of four women living in the northeastern United States, all with a diagnosis of NF1 predating an MS diagnosis; three of which were diagnosed with NF1 in infancy. The median age of diagnosis of MS was 42.5 years of age (range: 36–56 years of age). All patients in our cohort (4/4) had the presence of six or greater café-au-lait macules, cutaneous neurofibromas, and plexiform neurofibromas. Three out of four patients had skin fold freckling and Lisch nodules. Half had a family history of NF1. Three of the patients had both brain and spine lesions on imaging studies that had features more consistent with MS plaques rather than FASI often seen in people with NF1; the remaining patient only had lesions restricted to the subcortical white matter in the brain. All of our patients have a college degree or higher, two reported learning issues in childhood. The presenting symptoms for MS were variable in our cohort, yet the clinical scenarios included symptoms of MS commonly seen in people without NF1: vertigo, itchiness and paresthesia; dizziness; gait instability and bladder dysfunction. One of the patients was diagnosed based on incidental findings seen on an MRI of the brain that had been ordered by a provider outside of our system for an unclear indication.
At the data cutoff, all patients were neurologically stable with three of them receiving disease-modifying therapy and tolerating treatment well. The four patients are routinely monitored in our comprehensive NF1 center while their MS is managed largely by their primary neurologists.
Conclusion
Although rare, NF1 and MS can occur within the same patient. Previous literature has been inconsistent in the information reported about the patients with these concurrent diagnoses. Additional research is required to better understand the concurrent diseases and to determine if an underlying predisposition exists for NF1 patients. The few existing population studies looking at co-occurrence of NF1 and MS are limited by the large number of patients needed given the relative rarity of these diagnoses within the general population. Case reports and case series in the literature are often sporadic, and missing clinical data or treatment information. Translational research on the mechanism of co-occurrence is limited as well.
A logical question is how should we treat MS co-occurring with NF1 differently than MS alone, if at all? Newer-generation MS DMT such as B-cell directed monoclonal antibodies (e.g. ocrelizumab) are common and effective treatments in MS, however the risk for malignancy in those with underlying tumor predisposition syndromes such as NF1 is not yet understood.
To better characterize the dual disease states, prospective work is greatly needed. Gathering detailed clinical data, possibly through a patient registry, may be an avenue for future work and furthering our understanding. Consistent reporting of data will help characterize the dual disease processes, as well as additional treatment needs faced by these patients. Ultimately, these data would provide guidance for better treatment paradigms for patients with both MS and NF1.
Footnotes
Acknowledgments
This abstract was presented at the 2024 Global NF Conference in Brussels, Belgium.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Ethical considerations
Human and animal studies have been approved by the appropriate ethics committee and have therefore been performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments. For the case series, IRB waiver of HIPAA privacy authorization was obtained from the Johns Hopkins University School of Medicine (IRB 00403582). Individual participants for the case series additionally provided verbal informed consent for inclusion of their deidentified information and imaging.
Consent to participate
IRB waiver of HIPAA privacy authorization was obtained from the Johns Hopkins University School of Medicine (IRB 00403582). Individual participants for the case series additionally provided consent to participate.
Consent to publish
IRB waiver of HIPAA privacy authorization was obtained from the Johns Hopkins University School of Medicine (IRB 00403582). Individual participants for the case series additionally provided verbal informed consent for inclusion of their deidentified information and imaging. Each participant was given the opportunity to review the paper prior to submission.
Data availability
All pertinent data (lists of abstracts and articles, data entry) will be made available to qualified investigators on request. The abstract was previously presented at the 2024 Global NF Conference in Brussels, Belgium.