
Abstract
Background
Recombinant human immunoglobulin M22 (rHIgM22) has promoted remyelination in animal models and was well tolerated in people with clinically stable multiple sclerosis.
Objective
Safety/tolerability of a single rHIgM22 dose was investigated following an acute relapse and to determine whether this enhanced CNS/CSF concentrations.
Methods
Adults (N = 27) with acute relapse were assigned to rHIgM22 (0.5 or 2.0 mg/kg) or placebo. Study included screening/steroid administration periods and 10 study visits over 6 months. rHIgM22 CSF concentrations were assessed on days 2 and 29. Pharmacokinetic and safety samples were taken for up to 60 days. Assessments included adverse events and other clinical measures. Brain magnetic resonance imaging was performed with/without gadolinium.
Results
rHIgM22 CSF levels were consistent with dose-dependent concentration on both days 2 and 29. Infusion was generally well tolerated during an acute relapse. Immunogenicity was mild. Most adverse events did not appear to be dose dependent, were mild/moderate, and were events often associated with multiple sclerosis.
Conclusion
Although limited by high variability and small sample size, the data suggest enhanced CNS uptake associated with a drop in CSF levels. This study demonstrated safety of an antibody directed to myelin and oligodendrocytes in the course of active demyelinating disease. Further research into rHIgM22 is warranted.
Introduction
Current approved treatments for relapsing-remitting MS aim to minimize relapses, prevent disability, or limit the extent of injury during an acute relapse. However, there is no currently available treatment that directly promotes repair of myelin, so remyelinating therapies are a critical unmet need for the >2 million people worldwide who have the disease. 1
Recombinant human immunoglobulin M22 (rHIgM22) is a recombinant human-derived IgM, with human H and L chains and a murine J chain that promotes remyelination in animal models of MS.2–4 Preclinical data have shown that rHIgM22 binds to white matter in the central nervous system (CNS), and this binding starts a signal cascade that ultimately results in increased expression of myelin-related gene products. 5 rHIgM22 has been shown to promote remyelination in chronic virus–induced demyelinated3,6 and cuprizone-treated mice, and was well tolerated and showed beneficial effects on myelination.7–10 It also showed a significant acceleration in remyelination in animal experiments using lysolecithin injections in the spinal cord. 11 In murine and rat cell line studies, rHIgM22 has been shown to promote the differentiation of oligodendrocyte precursor cells to mature oligodendrocytes consistent with their role in remyelination, where their failure to differentiate and remyelinate axons is thought to be an important cause of the demyelination seen in MS.6,7,9,10
In a phase 1, double-blind, placebo-controlled, single-ascending-dose study of rHIgM22 in people with clinically stable MS, doses of 0.025 to 2.0 mg/kg were well tolerated and demonstrated dose proportionality in pharmacokinetics. There were minimal, infrequent, adverse events (AEs) attributed to rHIgM22: mainly headaches and mild infusion-type reactions (pruritus, arthralgia, flushing). Cerebrospinal fluid (CSF) drug concentration measurements suggested that there was dose-dependent crossing of the blood-brain barrier (BBB). CSF to plasma rHIgM22 concentration increased from 0.003% on day 2 for both 1.0 and 2.0 mg/kg to 0.056% and 0.586% for 1.0 and 2.0 mg/kg, respectively, on day 29. The study was not powered to show significant differences in disease outcomes. 12
In the current study, the safety and tolerability of a single dose of rHIgM22 was investigated in a phase 1 study of MS patients within 30 days of an acute relapse. The objective was to investigate whether the drug had greater access to the CNS compartment in the setting of a more open BBB associated with an acute relapse, thereby providing greater remyelinating benefit and hence a better clinical response. Although rHIgM22 is being developed as potential therapy in a presumed autoimmune disease, the potential of making acute MS worse because it is a natural autoantibody directed against myelin and oligodendrocytes was a concern. Thus, this study evaluated the safety, pharmacokinetics, pharmacodynamics, and potential immunogenicity of rHIgM22 in this patient population. The study was not powered to assess clinical efficacy, but measures of clinical status were obtained to determine whether the disease was worsened.
Methods
Study design, consent, and approvals
This was a phase 1, multicenter, double-blind, randomized, placebo-controlled, single-dose-escalation study in subjects with relapsing MS. Selection criteria were as follows: diagnosis of MS (McDonald 2010 criteria 13 ) age 18 through 75 years; and previous clinical acute relapse, defined as new or worsening neurological symptoms attributed to MS that lasted ≥24 h preceded by a stable or improving neurological state of ≥30 days and not associated with fever or infection. Neurological symptoms were considered to be any symptom reasonably attributed to an MS relapse (eg, changes in sensorimotor function). In addition, subjects had to have ≥1 new, identifiable, measurable, and active lesion in the brain via MRI after administration of gadolinium (Gd). A distinct correlation between the clinical syndrome and the MRI was not required per protocol. Exclusion criteria included subjects with primary progressive MS, various medical conditions or medication usage that would potentially impair safe participation or interpretation of trial results, initiation of various disease-modulating therapies within prespecified intervals, history of infusion reactions to biologics, or any contraindication to brain MRI.
Subjects were randomized 2:1 to a single dose of rHIgM22 or placebo. Two dose levels, 0.5 and 2.0 mg/kg, were tested sequentially in 2 separate cohorts each comprising 15 subjects. These doses were well tolerated in the previous clinical study. 12 Placebo was a sterile, preservative-free, aqueous solution of phosphate-buffered saline. Decision to complete a dose cohort and to escalate the dose in the next cohort was based on absence of dose-limiting toxicity in subjects treated with rHIgM22. If dose-limiting toxicity occurred, additional subjects could be enrolled in a dose-reduction step and a subsequent half-step increase. However, the maximum number of subjects exposed to rHIgM22, including replacement of subjects who discontinued study prematurely, was not to exceed 40.
Study included a screening and steroid administration period, a 3-day in-clinic dosing and observation period, and 10 follow-up study visits over a period of approximately 6 months (Figure 1). During the presentation and screening phase, subjects received a 5-day treatment with high-dose (1000 mg/d) oral or intravenous (IV) methylprednisolone. The 2 methylprednisolone regimens have been shown to be equivalent in clinical trials. 14 Subjects had a brain MRI, with and without Gd, to establish the presence of a new active brain lesion. All further screening evaluations were completed within 6 days after completion of methylprednisolone treatment. After administration of their assigned study medication on day 1, subjects remained hospitalized for 48 h after completion of the infusion. They were monitored for AEs and for changes in physical and laboratory test results by electrocardiogram telemetry during infusion and for 24 h afterward. After the first 2 subjects in each dose cohort were enrolled, if no treatment-related dose-limiting toxicity was observed within the first 7 days after infusion, the remaining 13 subjects in the cohort (9 active, 4 placebo) were administered study drug, with no more than 4 subjects treated on the same day. If no dose-limiting toxicity was observed in the entire cohort within 14 days following the infusion, the next cohort was enrolled in the same manner. Blood samples for pharmacokinetic and safety laboratory samples were taken at screening and on days 1, 2, 3, 8, 15, 29, and 60. Pharmacodynamic, exploratory biomarker and immunogenicity assessments were taken at various time points through day 180. Subjects were monitored for AEs, toxicity, vital signs, physical and neurological status via electrocardiograms, and laboratory test results. Particular attention was paid to the potential for worsening CNS inflammation, cytokine release syndrome, allergic reactions such as anaphylaxis, and immune complex disease induced by rHIgM22. Infusions were stopped if a hypersensitivity reaction occurred during the administration period, or if there was any significant clinical event.

Study design. MRI, magnetic resonance imaging; rHIgM22, recombinant human immunoglobulin M22.
Clinical and laboratory measurements
Clinical assessments included physical examination, vital signs, recording of AEs, 12-lead electrocardiogram and cardiac monitoring, and determination of the Expanded Disability Status Scale (EDSS) and individual Functional System Score. Laboratory evaluations included hematology, liver function, metabolic evaluation, and urinalysis. Immunological tests included cytokine profile assays (interferon-γ, tumor necrosis factor-α, and interleukin 6), complement (complement 3 and 4), total complement (CH50), and anti-rHIgM22 and antidrug antibody levels. Brain MRI was performed with and without Gd contrast. In addition, multimodal MRI tests that included magnetic resonance spectroscopy and magnetization transfer ratio (MTR) data were collected in a standardized fashion. Neurological findings were rated on the EDSS 15 including functional system scores. 16 Other efficacy measurements included Timed 25-Foot Walk (T25FW), 17 low contrast visual acuity, 18 pupillometry, 19 and Patient Global Impression of Change (PGI-C). 20
Statistical analyses
A sample size calculation was not performed for this study. It was anticipated that 30 subjects (20 assigned to active drug and 10 assigned to placebo) would be enrolled if there were no dropouts or dose-limiting toxicity. In the event of dropouts, and/or dose adjustments for dose-limiting toxicity, the maximum number of subjects exposed to active drug was not to exceed 40. Summary statistics and statistical analyses were performed using SAS® (SAS Institute Inc, Cary, NC) version 9.1.3 or higher. Dose dependence of rHIgM22 was tested via a nonparametric Jonckheere-Terpstra test. Pharmacokinetic parameters were estimated using noncompartmental methods with WinNonlin® (Pharsight Corp, Mountain View, CA). Correlations between rHIgM22 and albumin CSF concentrations and the Gd uptake index were fitted using locally estimated scatterplot smoothing regression and the Spearman rank correlation coefficient was calculated.
Results
All values are mean (SD) unless otherwise stated. The study was stopped owing to slow recruitment. The first subject was enrolled in August 2015, and the last subject completed the study in September 2018. A total of 27 subjects were randomized to rHIgM22 0.5 mg/kg (n = 10), rHIgM22 2.0 mg/kg (n = 9), and placebo (n = 8), in 10 sites in the United States before the study was terminated. No dose-limiting toxicity occurred. The study was completed by 18 of the 19 (94.7%) rHIgM22 subjects, and all of the 8 placebo subjects. One subject who enrolled in the 0.5-mg/kg rHIgM22 cohort withdrew owing to an acute infusion reaction and hypotension; hence, the pharmacokinetic population of rHIgM22 consisted of 18 subjects (0.5 mg/kg [n = 9] and 2.0 mg/kg [n = 9]). The safety population comprised all randomized subjects (n = 27). Demographic and baseline characteristics of the safety population are shown in Table 1. Subjects were mainly white and female with a mean age of 35.9 (10.0) years. The enrolled ratio (approximately 1:4 male to female) is similar to the MS prevalence sex difference
Demographics and baseline characteristics (safety population).

BMI, body mass index; EDSS, Expanded Disability Status Scale; MS, multiple sclerosis; rHIgM22, recombinant human immunoglobulin M22; T25FW, Timed 25-Foot Walk.
Safety
There were 121 AEs reported in 19 (100%) of subjects for the combined rHIgM22 groups and 50 were reported in 6 (75%) of subjects in the placebo group. Distribution of treatment-related or possibly treatment-related AEs and distribution of serious AEs considered treatment related was similar across groups (Table 2). One subject experienced an acute infusion reaction (serious AE), considered probably related to rHIgM22. The infusion was stopped at the onset of the reaction, treated appropriately, and no further dose was administered. This subject continued on the study until withdrawing consent before completion. There were no other discontinuations due to AEs and there were no deaths. Most treatment-emergent AEs (TEAEs) were mild or moderate in severity, and occurrences of these events did not appear to be dose dependent. The most common AEs in the rHIgM22 groups were headache in 12 out of 19 subjects (63.2%): back pain (5 subjects, 26.3%); post lumbar-puncture headache (4 subjects, 21.1%); and nausea, vomiting, dizziness, and oropharyngeal pain (3 subjects, 15.8% each). Pain in extremity, upper respiratory tract infection, urticaria, flushing, seasonal allergy, insomnia, and tachycardia occurred in 2 subjects (10.5%) each. All other TEAEs occurred in no more than 1 subject each. The most common AEs in the placebo group occurring in more than 1 subject were urinary tract infection in 3 subjects (25%), and malaise and nasopharyngitis in 2 subjects (25%). There were no TEAEs rated as Common Terminology Criteria for Adverse Events score ≥3 (severe, life threatening/disabling, or fatal). Four subjects (1 placebo [12.5%] and 3 rHIgM22 [15.8%]) experienced 7 infusion-related TEAEs (eg, infusion site pain, erythema, pruritus, edema, bruising) that were either mild or moderate in severity and all recovered. The 4 serious AEs in the rHIgM22 group were vomiting, acute infusion-related reaction, neck pain, and arthralgia. Each occurred in 1 subject. Only 1 of these, acute infusion reaction, was considered probably related to treatment. In the placebo group, 1 subject experienced multiple new CNS lesions that required hospitalization, which was considered by the investigator to be possibly related to the study drug before the drug status was revealed during a safety review.
Adverse events.

AE, adverse event; rHIgM22, recombinant human immunoglobulin M22.
Classified as “related” or “possibly related.”
“Procedural headache” AEs represent those events thought to be related to lumbar puncture. Otherwise headaches are recorded as “headaches” as per the protocol.
Anti-therapeutic antibodies
Anti-therapeutic antibodies increased from baseline over approximately 60 days and then declined (Table 3). The impact of drug-binding antibodies in this cohort is unclear.
Antitherapeutic (anti-rHIgM22) antibody immunogenicity by visit.

rHIgM22, recombinant human immunoglobulin M22.
Pharmacokinetics
Plasma concentrations are shown in Figure 2. Pharmacokinetic parameters are shown in Table 4 and CSF boxplots in Figure 3. CSF levels of rHIgM22 were consistent with a dose-dependent concentration on both days. On day 2, mean rHIgM22 CSF concentrations were 0.75 ng/mL (1.31) and 1.03 ng/mL (0.50) for the 0.5- and 2.0-mg/kg doses, respectively, and 0 and 0.08 ng/mL (0.09), respectively, on day 29. The half-life of rHIgM22 in mice is 15.4 h 3 and in normal mice rHIgM22 reaches the brain and spinal cord at a maximum of 0.3% of circulating dose at 24 h after intraperitoneal delivery. The P values for dose dependency were 0.024 for day 2 and 0.012 for day 29 via the nonparametric Jonckheere-Terpstra test Mean ratios of concentration in CSF to plasma on day 2 were 1.2 × 10−4 and 0.41 × 10−4 for the 0.5 and 2.0 mg/kg cohorts, respectively. On day 29, mean ratios were 0 and 10.9 × 10−4, respectively. Interpretation of CSF data is limited by high variability and small sample size. No rHIgM22 was detected in the CSF of placebo-treated patients.

Mean plasma concentration (SD) of rHIgM22 over 60 days. rHIgM22, recombinant human immunoglobulin M22.

Box plots of CSF rHIgM22 Cmax and AUC0−last AUC0−last, area under the concentration-time curve from time 0 to the concentration at last time point; Cmax, maximum measured concentration; CSF, cerebrospinal fluid; rHIgM22, recombinant human immunoglobulin M22.
Pharmacokinetic parameters for rHIgM22 (pharmacokinetic population).

AUC0-last, area under the concentration-time curve from time 0 to the concentration at last time point; CL, clearance; Cmax, maximum measured plasma concentration; CV, coefficient of variation; rHIgM22, recombinant human immunoglobulin M22; t½, half-life; tmax, time to maximum plasma concentration; Vz, terminal volume of distribution.
Pharmacodynamics
Mean baseline EDSS scores for placebo and all rHIgM22-treated subjects were 3.1 (1.27) and 3.1 (1.26), respectively. Subjects, including those receiving placebo, showed improvement in EDSS scores at postinfusion days 90 and 180 relative to baseline, although rHIgM22 did not show statistically significant improvement over placebo. Mean time to trial completion of the T25FW assessment at baseline showed comparable baseline values (4.0 ft/s [1.18] placebo; 4.6 ft/s [0.88] 0.5 mg/kg; 4.2 ft/s [0.81] 2.0 mg/kg; and 4.4 ft/s [0.85] overall rHIgM22). Subjects in all cohorts showed an upward trend in the speed with which they completed the T25FW. rHIgM22-treated subjects trended upward to a maximum speed between day 15 (5.0 ft/s [1.24]) and day 29 (4.9 ft/s [1.08]), then a downward trend to values approximating baseline by day 180. No meaningful conclusion regarding the effect of rHIgM22 dose or postinfusion time could be drawn.
Low contrast visual acuity and pupillometry and Patient Global Impression of Change
There were no clinically meaningful differences observed across treatments for visual acuity and pupillometry. PGI-C changes were also not significantly different between cohorts; for example, at day 90 there were 11/19 (57.9%) subjects receiving rHIgM22 who reported they were “moderately better,” “better,” or “a great deal better” compared with 4/8 (50.0%) of placebo subjects. The PGI-C results as shown by the visual analogue scale also showed no significant difference between cohorts.
Magnetization transfer ratio
The mean whole-brain median MTR at baseline was comparable across the rHIgM22 cohorts and the placebo cohort. Baseline mean values were 0.5 (0.29) for placebo and 0.4 (0.40) for rHIgM22 cohorts combined. Change from baseline to day 180 was −0.017 (0.09) in the rHIgM22 cohorts and −0.074 (0.08) in the placebo cohort. Postlesion MTR recovery least squares mean difference showed nonsignificant changes for the rHIgM22 cohorts vs placebo: 0.174 for 0.5 mg/kg (P = 0.315) and −0.233 (P = 0.139) for 2.0 mg/kg. High variability and small sample sizes preclude drawing any conclusion from these data.
New lesions were detected by MTR in 14 subjects overall over the course of the study: 4 in the placebo cohort and 10 in the rHIgM22 cohorts. A total of 46 new lesions were observed: 13 lesions in 4 (50%) subjects in the placebo group, 16 lesions in 6 (60%) subjects in the 0.5-mg/kg group, and 17 lesions in 4 (44%) subjects in the 2.0-mg/kg group. There was no indication that rHIgM22 treatment was worsening the disease.
Correlation between rHIgM22 CSF concentrations and brain Gd uptake
A significant negative correlation was observed between CSF concentration of rHIgM22 and the Gd uptake index for the 0.5-mg/kg rHIgM22 dose at day 2 (Figure 4A). A moderate negative correlation was also observed for 2.0 mg/kg at day 2 (Figure 4B). A plot of the CSF/serum albumin index vs the Gd uptake index for day 2 showed a plot that trended upward for albumin together with the Gd uptake index for both rHIgM22 doses, although there was no significant correlation (Figure 4C). The small sample size did not allow for a regression analysis for the CSF concentration rHIgM22 0.5-mg/kg dose vs Gd uptake index.
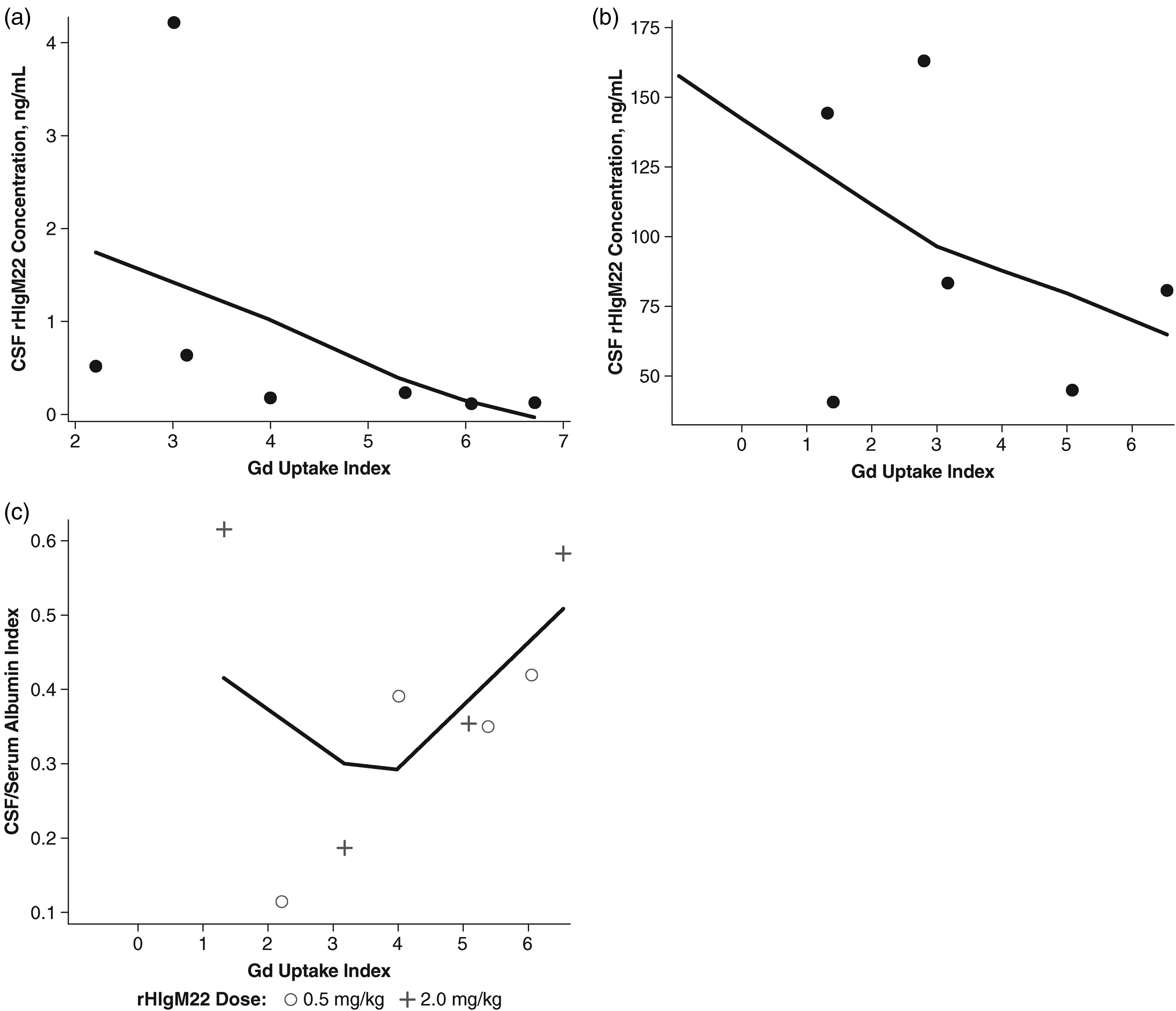
CSF rHIgM22 concentration and CSF/serum albumin index vs brain Gd uptake index at day 2. (a) CSF rHIgM22 concentration for 0.5-mg/kg rHIgM22 dose. Spearman r = −0.86 (P = 0.01). (b) CSF rHIgM22 concentration for 2.0-mg/kg rHIgM22 dose. Spearman r = −0.54 (P = 0.22). (c) CSF/serum albumin index for 0.5-mg/kg and 2.0-mg/kg rHIgM22 dose. The data were fitted using locally estimated scatterplot smoothing (LOESS) regression because the distribution was not normally distributed. CSF, cerebrospinal fluid; Gd, gadolinium; rHIgM22, recombinant human immunoglobulin M22.
Discussion
The main goal of this double-blind, placebo-controlled, single-ascending-dose IV infusion study of rHIgM22 in subjects with MS immediately following a relapse was to show that administration of rHIgM22 was safe and tolerable in those with active MS attacks. The selection criteria required participants to have clinical relapse and MRI evidence of a new brain lesion, but the number of new lesions observed during this study was surprisingly high (46 in 14 subjects) and unexpected—1 or 2 new lesions in patients on disease-modifying therapy is more typical—but this number of new lesions was similar in both treatment and placebo groups, indicating that they are likely not due to rHIgM22 treatment. Patients entering this study were treated with standard corticosteroids and were on disease-modifying therapies. Thus, despite these patients having very active disease, rHIgM22 autoantibody did not result in worsening of MS. The high rate of new lesions may suggest that the population was not optimized to detect improvement in clinical symptoms in this study.
Despite the number of new lesions, single infusions of rHIgM22 up to a maximum tested dose of 2.0 mg/kg appear to be safe and tolerable in subjects with MS. One patient in the treated arm and one patient in the placebo arm experienced a treatment-related serious AE. The infusion reaction experienced by the study drug–treated patient was managed effectively with standard protocols. In patients with acute relapses it would be anticipated that the breakdown in the BBB would coincide with an increase in Gd uptake and also uptake of the rHIgM22 antibody into the CSF. Interestingly, the data (shown in Figure 4) was the inverse of this expected result, with a lower observed rHIgM22 CSF concentration despite the increased porosity of the BBB. A hypothesis to explain this result is that rHIgM22 is absorbed into the brain in regions of active demyelination and therefore its concentration is reduced from what might be expected in the CSF. Mouse experiments in which labeled antibody was observed by MRI at sites of demyelination would support this conclusion.6,21,22 The flow of rHIgM22 from blood to brain to CSF has been disrupted as the rHIgM22 binds to cells or cellular elements within acutely, immunologically compromised myelin, thereby causing the negative correlation with the Gd uptake index (Figure 5). Notably, however, this theoretical explanation of the data has not been proven in this study. It is possible that there are unknown active processes that manage IgM penetration into the CSF compartment and further studies are warranted.

Hypothesis to account for pathway of rHIgM22 into the brain after an acute relapse. After an acute relapse, the BBB becomes more permeable, allowing large proteins (albumin and rHIgM22) to enter the CNS. The increase in albumin concentration is reflected in higher albumin concentrations in the CSF, with a positive correlation between albumin uptake index and Gd uptake index I. In contrast, rHIgM22 binds to white matter so does not remain in the CSF, resulting in a negative correlation with Gd uptake index. BBB, blood-brain barrier; CNS, central nervous system, CSF, cerebrospinal fluid; Gd, gadolinium; rHIgM22, recombinant human immunoglobulin M22.
Infusions were generally well tolerated, with mild injection site irritation reported primarily during the first 3 days following administration. The majority of safety observations were events often associated with MS. Immunogenicity, as detected by increased anti-rHIgM22 antibodies, was mild and occurred in about 35% to 40% of subjects over 2 months and declined to baseline levels after 6 months. The true impact of antitherapeutic and neutralizing antibodies, however, can only be determined in studies with larger numbers of participants. Plasma concentrations of rHIgM22 showed near linear dose proportionality of both maximum concentration (Cmax) and area under the concentration-time curve from time 0 to last time point (AUC0−last). Comparisons of plasma concentrations to drug concentrations in the CSF suggest that the amount of rHIgM22 that crossed the BBB was also dose dependent. The CSF concentrations observed here at day 2 in subjects after a relapse (0.7 ng/mL for the 0.5-mg/kg dose and 1.03 ng/mL for the 2.0-mg/kg dose) were comparable to those observed in patients with stable MS (with no evidence of active disease within 3 months) in the previous study at the same time after infusion (0.54 ng/mL for the 1.0-mg/kg dose and 1.13 ng/mL for the 2.0-mg/kg dose). 12
Exploratory endpoint findings were inconclusive, with no striking changes in EDSS scores, T25FW, pupillometry, or PGI-C identified for subjects receiving rHIgM22 vs those receiving placebo. The study was too small to reliably assess these changes.
Although this study was terminated early, and the small population size militated against any conclusions regarding clinical efficacy, this study importantly demonstrated safety in the course of very active MS. The hypothesis to explain the negative correlation between rHIgM22 CSF concentration and Gd uptake index may reflect its accumulation in the CNS and likely binding to the myelin target, which is of critical importance for a remyelinating antibody. Further research into rHIgM22 is warranted as a potential safe remyelinating treatment for both acute and chronic clinical deficits from MS.
Footnotes
Acknowledgments
This study was supported by Acorda Therapeutics, Inc. The sponsor of the study designed the study (with input from investigators), collected and analyzed the data, and was also involved in data interpretation and the decision to submit for publication. Editorial assistance was provided by Robin Smith, PhD, of The Curry Rockefeller Group, LLC (Tarrytown, NY), and this assistance was funded by Acorda Therapeutics, Inc (Ardsley, NY).
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: B.M. Greenberg has received consulting fees from Alexion, Novartis, EMD Serono, Viela Bio, Genentech/Roche, Greenwich Biosciences, Axon Advisors, Rubin Anders, ABCAM, Signant, IQVIA, Sandoz, Druggability Technologies, Genzyme, Immunovant, and PRIME Education. He has received grant funding from PCORI, NIH, NMSS, The Siegel Rare Neuroimmune Association, Clene Nanomedicine, and the Guthy Jackson Charitable Foundation for NMO. He serves as an unpaid member of the board of the Siegel Rare Neuroimmune Association. He receives royalties from UpToDate. J.D. Bowen has received compensation for activities such as advisory boards, lectures, and consultancy with the following companies and organizations: AbbVie, Alkermes, Biogen IDEC, Bristol Myers Squibb, Celgene, EMD Serono, Genentech, Genzyme, Novartis, and Roche. E. Alvarez has received compensation for activities such as advisory boards, lectures, and consultancy with the following companies and organizations: Actelion/Janssen, Alexion, Bayer, Biogen, Celgene/BMS, EMD Serono/Merck, Genentech/Roche, Genzyme, Novartis, and TG Therapeutics. He has also received research support from Biogen, Genentech/Roche, Novartis, TG Therapeutics, Patient-Centered Outcomes Research Initiative, National Multiple Sclerosis Society, National Institutes of Health, and Rocky Mountain MS Center. M. Rodriguez and A.E. Warrington: The Mayo Clinic owns patents and has licensed rHIgM22 to Acorda Therapeutics. If the antibody proves to be effective, then royalties may be received. A.O. Caggiano, Ping Zhao and A. Eisen were employees of Acorda Therapeutics at the time of the study, and A.O. Caggiano and A. Eisen own stock in Acorda.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Acorda Therapeutics, Inc (grant number N/A).