
Abstract
Background
PIRA (progression independent of relapse) has emerged as a term to quantify the proportion of disability worsening due to non-inflammatory neurodegenerative processes in multiple sclerosis (MS).
Objective
To determine the impact of PIRA on retinal thinning, a biomarker of neuroaxonal degeneration in MS, in comparison to traditional disability worsening and relapse.
Methods
In a 4-year, prospective observational study including 171 relapsing MS (RMS) patients, retinal thinning was determined by annual spectral-domain optical coherence tomography measuring macular ganglion-cell-and-inner-plexiform-layer (GCIPL) and peripapillary-retinal-nerve-fibre-layer (pRNFL). PIRA was defined as an expanded disability status scale (EDSS) or symbol digit modalities test (SDMT) worsening confirmed after 24 weeks with no relapse in the 30 days before or after the disability worsening.
Results
Each PIRA event was associated with a mean additional loss of GCIPL (1.8 µm) and pRNFL (1.9 µm), similar to the impact of EDSS and SDMT worsening. Overall relapse and relapse without subsequent EDSS worsening did not influence retinal thinning, while a relapse with EDSS worsening was associated with an additional loss of GCIPL (1.3 µm) and pRNFL (1.4 µm).
Conclusions
PIRA is associated with retinal thinning, likely reflecting neurodegenerative processes, not directly associated with focal inflammation. It might be a clinical measure to identify MS patients with ongoing MS-associated neurodegeneration.
Keywords
Introduction
Multiple sclerosis (MS) is a chronic autoimmune-mediated disease of the central nervous system (CNS) pathophysiologically comprising both inflammation and neurodegeneration. 1
The number of disease-modifying treatments (DMT) available for relapsing MS (RMS) is ever expanding. However, current DMTs are highly efficacious in suppressing inflammation (i.e. relapse rate, accumulation of new/enlarging T2 hyperintense [T2] and/or contrast-enhancing [CE] lesions on magnetic resonance imaging [MRI]), while efficacy on parameters attributed to neurodegeneration (i.e. disability worsening, brain atrophy determined by MRI) is much lower.
Evidence is growing that even in RMS patients treated with highly efficacious DMT, long-term disability worsening is not uncommon and associated with accelerated brain atrophy, but largely independent of relapse activity. 2
In recent years, a new term has emerged in MS research that is PIRA, or progression independent of relapse activity. 3 PIRA can be variously defined as worsening disability (with EDSS or a composite) independent of relapses (within a defined period or in relapse-free patients) and occurs in 10–25% of RMS patients over 4–5 years representing about 50–80% of all disability worsening events.3–6 Thus, PIRA is purported to quantify the proportion of disability worsening due to (non-inflammatory) neurodegenerative processes.
Optical coherence tomography (OCT) enables non-invasive and inexpensive in-vivo measurement of distinct layers of the retina. In MS, atrophy of the macular ganglion cell layer plus the adjacent inner plexiform layer (GCIPL) and the peripapillary retinal nerve fibre layer (pRNFL) are established markers of neurodegeneration in MS. GCIPL and pRNFL thickness correlate with and also predict physical and cognitive disability progression.7–10
Thus, the objective of this study was to determine the impact of PIRA on retinal thinning in comparison to traditional physical disability worsening and relapse.
Materials and methods
We included 171 patients diagnosed with RMS according to the 2010 McDonald criteria aged between 18 and 65 years from a prospective, observational study on OCT in RMS.8–10
Clinical study visits were conducted every three months for four years with demographic data, EDSS, neurological and treatment history including DMT obtained from each participant at every visit. EDSS worsening was defined as a confirmed EDSS increase of ≥1.0 point in patients with a baseline score of ≤5.5, or an increase of ≥0.5 points in patients with a baseline score of >5.5 sustained for at least 12 months as compared to baseline. 11 For assessment of cognitive dysfunction, the Symbol Digit Modalities Test (SDMT) was obtained annually, which is particularly suitable for longitudinal assessment of MS-related cognitive changes.12–14 SDMT worsening was defined as a loss of ≥4 points or a ≥10% decrease in SDMT score as compared to baseline sustained for at least 12 months. 14 A relapse was defined as objectively observed signs typical of an acute CNS inflammatory demyelinating event, current or prior to the visit, with duration of at least 24 hours in the absence of fever or infection, separated from the last relapse by at least 30 days. 15 Relapses were further classified as either resulting or not resulting in a subsequent EDSS worsening sustained 6 months after relapse (relapse with/without EDSS worsening) compared to the last EDSS before the relapse. PIRA was defined as either an EDSS or SDMT worsening during the observation period confirmed after 24 weeks with no relapse in the 30 days before or after the EDSS/SDMT worsening.3,5
DMT was grouped as following: 1) “no DMT” (N-DMT) defined as patients receiving no DMT at least 6 months prior to baseline visit and during the whole observation period, 2) “moderate efficacious DMT” (M-DMT) defined as patients receiving one or more DMT of either interferon beta preparations, glatiramer acetate, dimethylfumarate, or teriflunomide during the whole observation period, 3) “highly efficacious DMT” (H-DMT) defined as patients receiving one or more DMT of either natalizumab, fingolimod, alemtuzumab, ocrelizumab or cladribine during the whole observation period, and 4) “ESC-DMT” defined as patients in whom DMT was escalated either from no DMT to moderate or from moderate to highly efficacious DMT during the observation period.9,10
OCT
Spectral-domain OCT (Spectralis, Heidelberg Engineering, Heidelberg, Germany; software Spectralis Software Version 6.9a) was performed annually without pupil dilatation in a dark room on both eyes of each patient. For GCIPL measurement, a 20°×20° macular volume scan (512 A-scans, 257 B-scans, vertical alignment, automatic real time [ART] 16 frames) automatically centered around the fovea was done. GCIPL thickness was defined as the mean thickness of the inner four quadrants of the grid (corresponding to the 3-mm ring as defined by the Early Treatment Diabetic Retinopathy Study). 16
For pRNFL measurement, a custom 3.4 mm ring scan (12°) centred on the optic nerve head was used (1536 A-scans, ART 100). Image processing was conducted semiautomated with manual correction of obvious errors. All examinations were checked for sufficient quality using OSCAR-IB criteria. 17 OCT results are reported according to APOSTEL guidelines. 18
Thicknesses of mGCIPL and pRNFL were calculated as the mean of the values for both eyes. Patients with a history of unilateral ON <6 months before baseline were excluded from the study. Eyes with a history of ON more timely distant were not excluded as further retinal thinning does not differ between eyes with and without a history of optic neuritis. 7 Eyes suffering ON during the observation period were excluded from the study and only the values of eyes without ON during the observation period were used for calculation of retinal thinning in the analyses. To identify subclinical ON during the course of the study, we used interocular asymmetry in retinal thinning (i.e. inter-eye difference in mGCIPL/pRNFL thickness reduction compared to the prior OCT) with cut-off values of ≥4 µm for GCIPL and ≥5 µm for pRNFL.19,20 In these cases, only the eye with the higher value was analyzed. 17 Other exclusion criteria were previous diagnoses of ophthalmological (i.e. myopia greater than -4 diopters, optic disc drusen), neurological, or drug-related causes of vision loss or retinal damage not attributable to MS. 21 The investigators performing the OCT were blinded to clinical parameters and vice versa.
Statistical analysis
Statistical analysis was performed using SPSS 25.0 (SPSS Inc, Chicago, IL). Categorical variables were expressed in frequencies and percentages, continuous variables as mean and standard deviation (SD) or median and range as appropriate.
Annual thinning rates of GCIPL (annualised loss of GCIPL = aLGCIPL) and pRNFL (annualised loss of pRNFL = aLpRNFL) were determined by individual linear regression models as the slope of the regression line best fitted to all measurements over the observation period. To analyse the influence of PIRA, EDSS/SDMT worsening and relapse, we performed linear regression models with aLGCIPL/aLpRNFL as the dependent variable with stepwise adjustment for age, sex, disease duration, GCIPL/pRNFL at baseline, EDSS/SDMT at baseline and DMT. To investigate potential interaction between PIRA, EDSS/SDMT worsening and relapse in influencing retinal thinning, we calculated combined linear regression models including interaction terms. Missing values were handled by multiple (20 times) imputation using the missing not at random (MNAR) approach with pooling of estimates according to Rubin’s rules. 22 A two-sided p-value <0.05 was considered statistically significant.
Results
The inclusion process is depicted in detail in Figure 1. Characteristics of the final study cohort are given in Table 1.

Flow diagram.
Demographic and clinical characteristics of the cohort.

aNumber (percentage).
bMean and standard deviation.
cMedian and range. p-values calculated for comparison of stable and clinically progressing patients using.
dChi-square-test.
eIndependent t-test.
fMann-Whitney-U-test as appropriate.
DMT: disease modifying therapy. EDSS: Expanded Disability Status Scale. GCIPL: macular ganglion cell and inner plexiform layer. MS: multiple sclerosis. N-PIRA: no PIRA. PIRA: progression independent of relapse. pRNFL: peripapillary retinal nerve fibre layer. SDMT: Symbol Digit Modalities Test. N-DMT: patients receiving no DMT at least 6 months prior to baseline visit and during the whole observation period. M-DMT: defined as patients receiving one or more DMT of either interferon beta preparations, glatiramer acetate, dimethylfumarate, or teriflunomide during the whole observation period. H-DMT: defined as patients receiving one or more DMT of either natalizumab, fingolimod, alemtuzumab, ocrelizumab and cladribine during the whole observation period. ESC-DMT: defined as patients in whom DMT was escalated either from N-DMT to M-DMT or from M-DMT to H-DMT during the observation period. NA: not applicable.
PIRA occurred in 41 (24.0%) patients. There were no significant differences in demographics and baseline clinical characteristics between the PIRA and the no PIRA (N-PIRA) groups (Table 1). Mean pRNFL and GCIPL thicknesses at baseline were significantly lower in the PIRA group compared to the N-PIRA group.
Mean annual retinal thinning was 1.3 µm (SD 1.7) for pRNFL and 1.0 µm (SD 1.4) for mGCIPL in the whole cohort. The slopes of change in retinal thinning were significantly steeper in patients with EDSS or SDMT worsening, but also in patients presenting PIRA (Figure 2(a) to (d), (i) and (j)). While there was no significant difference in retinal thinning between patients with and without relapse activity during the observation period (Figure 2(e) and (f)), patients suffering a relapse resulting in subsequent EDSS worsening displayed steeper decline in both pRNFL and GCIPL in comparison to relapse-free patients or with relapse without EDSS worsening (Figure 2(g) and (h)). Frequency of PIRA events did not significantly differ between patients receiving no DMT (0.03 per year), M-DMT (0.02/year), H-DMT (0.02/year) or ESC-DMT (0.04/year) during the observation period.
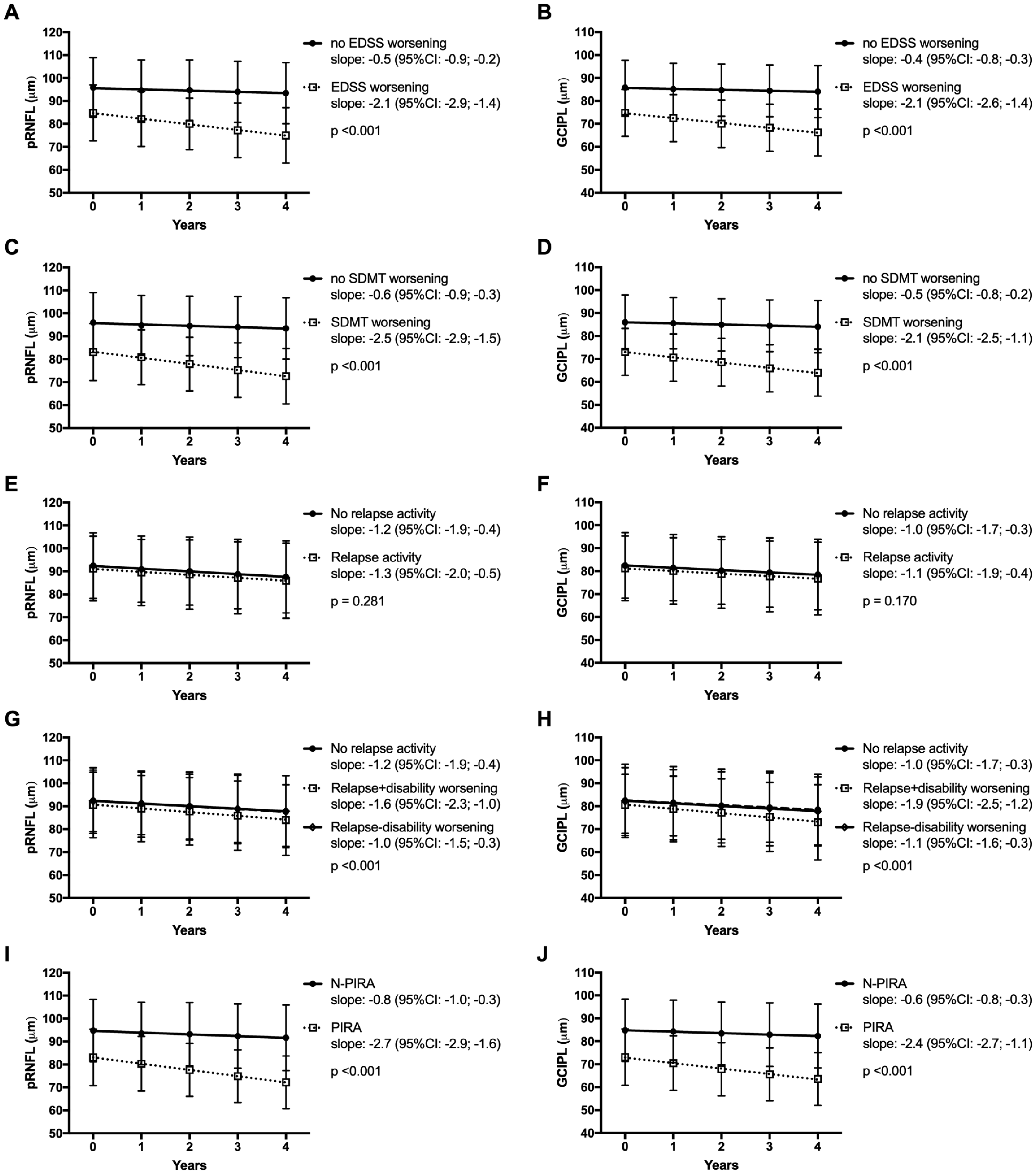
Change of pRNFL and GCIPL depending on EDSS worsening (a, b), SDMT worsening (c, d), relapse activity (e, f), Relapse with and without disability progression (g, h) and progression independent of relapse (i, j).
Next, we analysed the influence of clinical events on retinal thinning by multivariate regression analysis adjusted for age, sex, disease duration, baseline characteristics (EDSS, SDMT, GCIPL/pRNFL thickness) and DMT status during the observation period. Each PIRA event was associated with a mean additional loss of 1.8 µm in GCIPL and 1.9 µm in pRNFL (Table 2). The impact of EDSS and SDMT worsening was similar to PIRA. In the combined model, there was significant interaction between the effect of PIRA and EDSS/SDMT worsening on retinal thinning (p < 0.001, respectively). While a single relapse event without distinction regarding subsequent EDSS worsening was not significantly associated with retinal thinning, a relapse with EDSS worsening resulted in an additional loss of GCIPL (1.3 µm) and pRNFL (1.4 µm), while a relapse without EDSS worsening was not.
Annual loss of pRNFL and GCIPL depending on disability worsening, relapse and progression independent of relapse.

aLpRNFL: annualised loss of peripapillary retinal nerve fibre layer. aLGCIPL: annualised loss of macular ganglion cell and inner plexiform layer. B: regression coefficient. 95% CI: 95% confidence interval for B. EDSS: Expanded Disability Status Scale. PIRA: progression independent of relapse activity. SDMT: symbol digit modalities test.
aValues indicate an additional loss of B µm in pRNFL/GCIPL per occurrence of the respective event calculated by linear regression models.
Discussion
In this study we aimed to determine the association of retinal thinning, an established marker of neuroaxonal loss in MS, with progression independent of relapse (PIRA), a term suggested to reflect the proportion of disability worsening caused by diffuse neurodegenerative processes not primarily caused by focal inflammation, in comparison to traditional disability worsening and relapse.
We found that PIRA has a significant impact on retinal thinning with an effect size very similar to traditional definitions of disability worsening such as EDSS and SDMT worsening. Importantly, relapses did not influence retinal thinning when no distinction was made regarding the relapse resulting in subsequent EDSS worsening. However, when relapses were subclassified as relapse with subsequent EDSS worsening, they were associated with an additional loss of GCIPL and pRNFL, whereas a relapse without subsequent EDSS worsening did not affect retinal thinning.
PIRA is an evolving concept, similar to “silent progression”, another recently emerging term, implying a discrimination between disability worsening not associated to relapse as opposed to disability worsening in the wake of relapse. Its aim is to quantify the proportion of disability worsening due to MS associated neurodegenerative processes. While the underlying pathophysiology is unclear, MS associated neurodegeneration is likely not driven by acute focal inflammation, which clinically corresponds to the event of acute relapse, but rather by diffuse injury caused by chronic inflammation or possibly even non-inflammatory processes, which is clinically reflected by disability worsening independent of relapses. Evidence indicating that disability worsening, which occurs independently of relapses or white-matter lesions, is correlated with brain atrophy strengthens the plausibility of this concept. 2
Retinal thinning in absence of optic neuritis is a robust and established marker of MS-associated neuroaxonal damage correlated with disability worsening and faster rates of whole brain, thalamic, and grey matter atrophy, but independent of relapse activity.7,8,10,23,24 Our results clearly show that PIRA events are reflected by retinal thinning adding an important component of evidence to the concept of PIRA. However, the very important question of which comes first or who is driving whom, retinal thinning or disability worsening, remains to be elucidated.
The proportion of patients displaying PIRA events in our cohort was 24% over 4 years comprising 59% of all disability worsening events which is well within the range of previously reported rates of PIRA.3,4,6 Thus, a high proportion of disability worsening events occur in the absence of relapses even in RMS patients treated with high efficacious DMT, although the number of PIRA events in absolute terms is low in RMS (perhaps 2-5%/year). 2
Of note, the frequency of PIRA events was similar between DMT-groups ranging from 2-4% per year in our cohort which is comparable to results of other studies.2,4 Consequently, PIRA might represent the stratum of MS-associated neurodegeneration that is largely unaffected by DMTs predominantly targeting CNS inflammation.3,4,6 This evidently shows that disability worsening independent of relapses is by no means restricted to patients diagnosed with progressive MS (PMS), ultimately questioning the usefulness of the dichotomous diagnostic distinction in patients with RMS and PMS. 25 It might be more reasonable to label MS patients in a dual-axis, multimodal system as i) currently showing signs of acute focal inflammatory activity or not (relapses, new/enlarging T2 or CE lesions) and ii) currently showing signs of ongoing MS-associated neurodegeneration (physical or cognitive disability worsening independent of relapses, brain atrophy, retinal thinning) or not.
However, the applied definitions of PIRA have been varying both regarding disability worsening (using either EDSS or a composite) and independency of relapses (either a defined period of time between relapse and disability worsening or only relapse-free patients). In our cohort, using a composite of EDSS and SDMT for defining disability worsening and a defined period unsurprisingly yielded a proportion of PIRA at the upper range of the reported spectrum. Like other composite metrics (such as no evidence of disease activity, NEDA), PIRA requires a standardizing of its definition and methodology in order to enable reliable comparability and interpretability of results. 26 It may benefit from including MRI parameters of inflammatory activity such as new T2 or CE lesions to unveil short-term disability worsening due to acute focal inflammation. Unfortunately, we did not have MRI or body fluid biomarkers available for correlation. In this context, it is noteworthy that not all events classified as relapses are really associated with focal neuroinflammatory events. In our study, this potential bias is minimized by meticulously following the relapse definition. 15 Additionally, we only accepted events as relapses where objectifiable signs typical of an acute CNS inflammatory demyelinating event were documented as opposed to solely patient reported symptoms.
As a limitation, we did not have sufficient data available to differentiate the impact of relapse on retinal thinning according to affected clinical functional systems or neuroanatomical localization.
In conclusion, PIRA is associated with retinal thinning, likely reflecting neurodegenerative processes, not directly associated with focal inflammation. When methodology is further refined, PIRA might be a clinical measure to identify MS patients with signs of ongoing MS-associated neurodegeneration complementing paraclinical measures such as retinal thinning.
Footnotes
Authors’ contributions
Gabriel Bsteh: study concept and design, patient recruitment, acquisition of data, statistical analysis and interpretation of data. Harald Hegen: patient recruitment, acquisition of data, interpretation of data, critical revision of manuscript for intellectual content. Patrick Altmann: critical revision of manuscript for intellectual content. Michael Auer: patient recruitment, acquisition of data, critical revision of manuscript for intellectual content. Klaus Berek: patient recruitment, acquisition of data, critical revision of manuscript for intellectual content. Franziska Di Pauli: patient recruitment, acquisition of data, critical revision of manuscript for intellectual content. Sebastian Wurth: patient recruitment, acquisition of data, critical revision of manuscript for intellectual content. Anne Zinganell: patient recruitment, acquisition of data, critical revision of manuscript for intellectual content. Paulus Rommer: critical revision of manuscript for intellectual content. Florian Deisenhammer: patient recruitment, acquisition of data, critical revision of manuscript for intellectual content. Fritz Leutmezer: critical revision of manuscript for intellectual content. Thomas Berger: study concept and design, patient recruitment, interpretation of data, critical revision of manuscript for intellectual content, study supervision.
Acknowledgements
The authors want to explicitly thank Yvonne Wehle and Daniela Schneider who diligently performed OCT scans for this study.
Conflict of Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Gabriel Bsteh: has participated in meetings sponsored by, received speaker honoraria or travel funding from Biogen, Celgene, Merck, Novartis, Sanofi-Genzyme and Teva, and received honoraria for consulting Biogen, Roche and Teva. Harald Hegen: has participated in meetings sponsored by, received speaker honoraria or travel funding from Bayer, Biogen, Merck, Novartis, Sanofi-Genzyme, Siemens and Teva, and received honoraria for consulting Biogen and Teva. Patrick Altmann: has participated in meetings sponsored by, received speaker honoraria or travel funding from Biogen, Merck, Roche, Sanofi-Genzyme and Teva, and received honoraria for consulting from Biogen. He received a research grant from Quanterix International and was awarded a combined sponsorship from Biogen, Merck, Sanofi-Genzyme, Roche, and Teva for a clinical study. Michael Auer: received speaker honoraria and/or travel grants from Biogen, Merck, Novartis and Sanofi Genzyme. Klaus Berek: has participated in meetings sponsored by and received travel funding from Roche. Franziska Di Pauli has participated in meetings sponsored by, received honoraria (lectures, advisory boards, consultations) or travel funding from Bayer, Biogen, Merck, Novartis, Sanofi-Genzyme, Teva, Celgene and Roche. Sebastian Wurth: has participated in meetings sponsored by, received honoraria or travel funding from Allergan, Biogen, Ipsen Pharma, Merck, Novartis, Roche, Sanofi Genzyme and Teva. Anne Zinganell: has participated in meetings sponsored by, received speaking honoraria or travel funding from Biogen, Merck, Sanofi-Genzyme and Teva. Paulus Rommer: has received honoraria for consultancy/speaking from AbbVie, Allmiral, Alexion, Biogen, Merck, Novartis, Roche, Sandoz, Sanofi Genzyme, has received research grants from Amicus, Biogen, Merck, Roche. Florian Deisenhammer: has participated in meetings sponsored by or received honoraria for acting as an advisor/speaker for Alexion, Almirall, Biogen, Celgene, Merck, Novartis, Roche and Sanofi-Genzyme. His institution received scientific grants from Biogen and Sanofi-Genzyme. Fritz Leutmezer: has participated in meetings sponsored by or received honoraria for acting as an advisor/speaker for Bayer, Biogen, Celgene, MedDay, Merck, Novartis, Roche, Teva and Sanofi-Genzyme. Thomas Berger: has participated in meetings sponsored by and received honoraria (lectures, advisory boards, consultations) from pharmaceutical companies marketing treatments for MS: Allergan, Bayer, Biogen, Bionorica, Celgene, MedDay, Merck, Novartis, Octapharma, Roche, Sanofi-Genzyme, Teva. His institution has received financial support in the past 12 months by unrestricted research grants (Biogen, Bayer, Merck, Novartis, Sanofi Aventis, Teva and for participation in clinical trials in multiple sclerosis sponsored by Alexion, Bayer, Biogen, Merck, Novartis, Octapharma, Roche, Sanofi-Genzyme, Teva.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.