
Abstract
Multiple sclerosis (MS) and systemic lupus erythematous (SLE) are autoimmune diseases, the coexistence of which is uncommon in patients. Owing to the rarity of this condition, the distinction between MS and SLE is a diagnostic challenge for neurologists. We present a case report in which MS and SLE were present in the same patient. There are few case reports in the world on the association between MS and SLE. The following case report is the first of its kind in which both MS and SLE are present in a patient from a country with low prevalence of MS such as Ecuador.
Background
Both multiple sclerosis (MS) and systemic lupus erythematous (SLE) are autoimmune diseases. 1 MS is caused by immune cell infiltration across the blood-brain barrier, which promotes inflammation, demyelination, gliosis and neuroaxonal degeneration of the white matter in the central nervous system (CNS).2,3 SLE is a B-cell-mediated autoimmune disease characterized by the generation of autoantibodies against nuclear antigens and a type III hypersensitivity leading to chronic systemic inflammation and tissue damage of various organs and systems.1,4 Genetic and environmental factors could have a role in the development of these diseases but the etiology is still unknown. 1 The presence of both diseases in the same patient is rare, which suggests a relative incompatibility between these diseases. 5
The prevalence of MS in the world is not homogeneous. It is higher in North American and European countries (> 100 per 100,000 inhabitants) than in South American countries such as Ecuador, in which the prevalence of MS is 1.2 per 100,000 inhabitants. 6 The prevalence of SLE varies considerably worldwide. In North America, Europe and Asia the prevalence is 52, 28–71 and 30–60 cases per 100,000 inhabitants, respectively. Thus far, however, there have been no epidemiological studies on the prevalence of SLE in Ecuador.
The diagnosis of MS in a patient with SLE can be a challenge as the association between MS and SLE is rare, and at present, only 17 case reports have been written in the world. Fanouriakis et al. 2 have reported the largest number of case reports with nine patients and in Latin America only one case report has been written. 7 For this reason, this case report will be the second in Latin America and the first in Ecuador.
Case report
We present a 35-year-old woman with no relevant previous diseases or family history of autoimmune disorders. At the age of 32, the patient was diagnosed with relapsing–remitting MS (RRMS) according to McDonald 2010 diagnosis criteria. 8 This diagnosis was established after paresis and numbness in her right foot and urinary retention was present at age 27, optic neuritis (ON) at age 29 and finally right hemiparesis at age 31. At the time of her diagnosis with RRMS, cerebrospinal fluid (CSF) oligoclonal bands (OCBs) were present. Notably, serum immunological tests, including serum antinuclear antibodies (ANA), anti-double-stranded DNA (anti-dsDNA) antibodies, anti-Ro/SSA and anti-La/SSB antibodies, antiphospholipid antibodies (aPL), human immunodeficiency virus (HIV) and Venereal Disease Research Laboratory (VDRL) tests were negative. Also, lactate dehydrogenase (LDH) and vitamin B12 levels were normal. Brain magnetic resonance imaging (MRI) showed multiple focal T2-weighted periventricular, juxtacortical and cerebellar hyperintensities. A spinal cord MRI showed one thoracic gadolinium-enhanced lesion (Figure 1). The Expanded Disability Status Scale (EDSS) was 2. The differential diagnosis of this case was conducted through the search for clinical and paraclinical red flags, which in this case were not present.

Brain magnetic resonance imaging (MRI). Axial fluid-attenuated inversion recovery image reveals plaques of demyelination in the white matter (a); one left juxtacortical lesion is seen (b). Spinal cord MRI. An enhancement lesion is seen in the right lateral white matter of the spinal cord ((c) and (d)). All lesions are indicated by arrows.
The patient had been treated with intravenous methylprednisolone for MS relapses resulting in complete recovery. At age 32, the patient started treatment with subcutaneous interferon (INF)-beta-1a, which she received for three years and no evidence of disease activity was seen. At age 35, the patient chose to stop treatment with IFN as she desired to become pregnant. Six months later, the patient was diagnosed with dengue and recovered completely within a week. However, one week after her recovery from dengue, she complained of asthenia, myalgia, arthralgia, hematuria, and fever. Generalized adenopathies were found in the physical exam and laboratory investigations showed anemia (hemoglobin 9.9 g/l), leukopenia (3.5 × 109/l), lymphopenia (0.68 × 109/l), positive direct Coombs and high LDH (666 U/ml) levels. Other laboratory findings, including renal function tests and thyroid hormones, were normal. However, proteinuria (300 mg/24 h) and C3 and C4 low complement levels were identified. Virologic tests were normal or negative, ANA titers were > 7.5 U/ml (<1.2 U/ml) and anti-dsDNA titers were 1/320 (<1/10). Anti-Ro/SSA antibodies were negative. Echocardiogram showed a mild, pericardial effusion. Based on the existence of hemolytic anemia, lymphopenia, leukopenia, serositis, positive ANA and anti-dsDNA antibodies, the diagnosis of SLE was established according to the American College of Rheumatology revised criteria for the classification of SLE. The patient started treatment with hydroxychloroquine and prednisone, which were prescribed by a rheumatologist, and a new brain MRI did not show new T2 or enhancing lesions. After three months, an SLE reactivation was diagnosed after an increase of proteinuria (2.9 gr/24 h) and complement consumption was observed. Following this diagnosis, a kidney biopsy was performed that showed proliferative glomerulonephritis with class III sclerotic lesions. As a result of the findings, a multidisciplinary evaluation by neurologists, nephrologists and rheumatologists was carried out. The consensus reached was that (a) the patient had active SLE, (b) the MS was inactive from the beginning of treatment with IFN despite treatment suspension for a period of nine months, (c) rituximab was the treatment to be administered and (d) there would be monthly follow-ups of the SLE for three months, after which there would be a trimonthly follow-up and in the case of MS, after six months of rituximab administration, a cerebral MRI would be performed. After the above-mentioned six months, no further neurologic relapse symptoms were noted and brain MRI did not show any additional lesions compared with images taken from the patient at age 32 and SLE was quiescent.
Discussion
In this case report, it is important to determine if the neurologic manifestations and the brain and spinal cord lesions in MRI were due to SLE, or a result of RRMS with post-development of typical systemic manifestations of SLE due to the fact that neurological manifestations in SLE can be present years before the systemic manifestations. 9 In SLE, aPL play a crucial role; the mechanism by which these antibodies can produce a disease similar to MS in patients with SLE includes the molecular mimetic with myelin, vasculopathy and autoimmune vasculitis. 7 However, in our patient, aPL was negative at the beginning of the disease and during the systemic manifestation of SLE.
ON can be present in MS and SLE. In MS, ON is characterized by an acute or subacute course, with unilateral or bilateral impairment of vision and retro-orbital or ocular pain that is usually exacerbated by eye movement; a total or partial recovery follows these clinical characteristics. ON in SLE is rare; however, the characteristic of ON is an acute visual impairment that is followed by progressive visual loss lasting weeks after the initial visual impairment. 10
Myelitis in MS is asymmetrical, progresses in hours or days and sphincter impairment is usually present. Myelitis in SLE is usually the first neurologic manifestation in around 21% of cases. Extensive longitudinal spinal cord damage is seen in 71% of patients and spinal cord swelling is seen in 91.7% of cases when myelitis affects gray matter. In SLE, there is a clear association between myelitis and lupus anticoagulants, both of which were negative in our patient. 11

MS: multiple sclerosis; SLE: systemic lupus erythematous; DIS: dissemination in space; ANA: antinuclear antibodies; CSF: cerebrospinal fluid.
In our case, the diagnosis of MS was based on the McDonald 2010 diagnosis criteria, which do not consider the presence of OCBs for the diagnosis of RRMS. 8 Our patient met criteria for dissemination in time and space (DIS) despite having positive OCBs, which, at that time, was not taken into consideration in the diagnosis. However, in recent years, OCBs have begun to play a fundamental role in patients with clinically isolated syndrome (CIS) and MS. 13 In this regard, a meta-analysis has shown that the presence of OCBs in patients with CIS predicts the conversion to clinically defined MS (CDMS) and this meta-analysis showed that the presence of OCBs in patients with MS was an indicator of disability progression measured by EDSS. 14 A prospective study in 415 patients with CIS showed that the presence of OCBs was associated with the conversion to CDMS, and the presence of OCBs increased the risk of a second relapse. 15 Arrambide et al. demonstrated that the presence of OCBs together with DIS could be an additional criterion for the diagnosis of MS in patients with CIS, which allowed the OCBs to be considered in the new McDonald 2017 diagnostic criteria.13,16 For this reason, we recommend testing for OCBs in patients with CIS since the presence of OCBs allows an earlier MS diagnosis and could be a useful predictor of disability.
Clinical characteristics of SLE-MS patients.
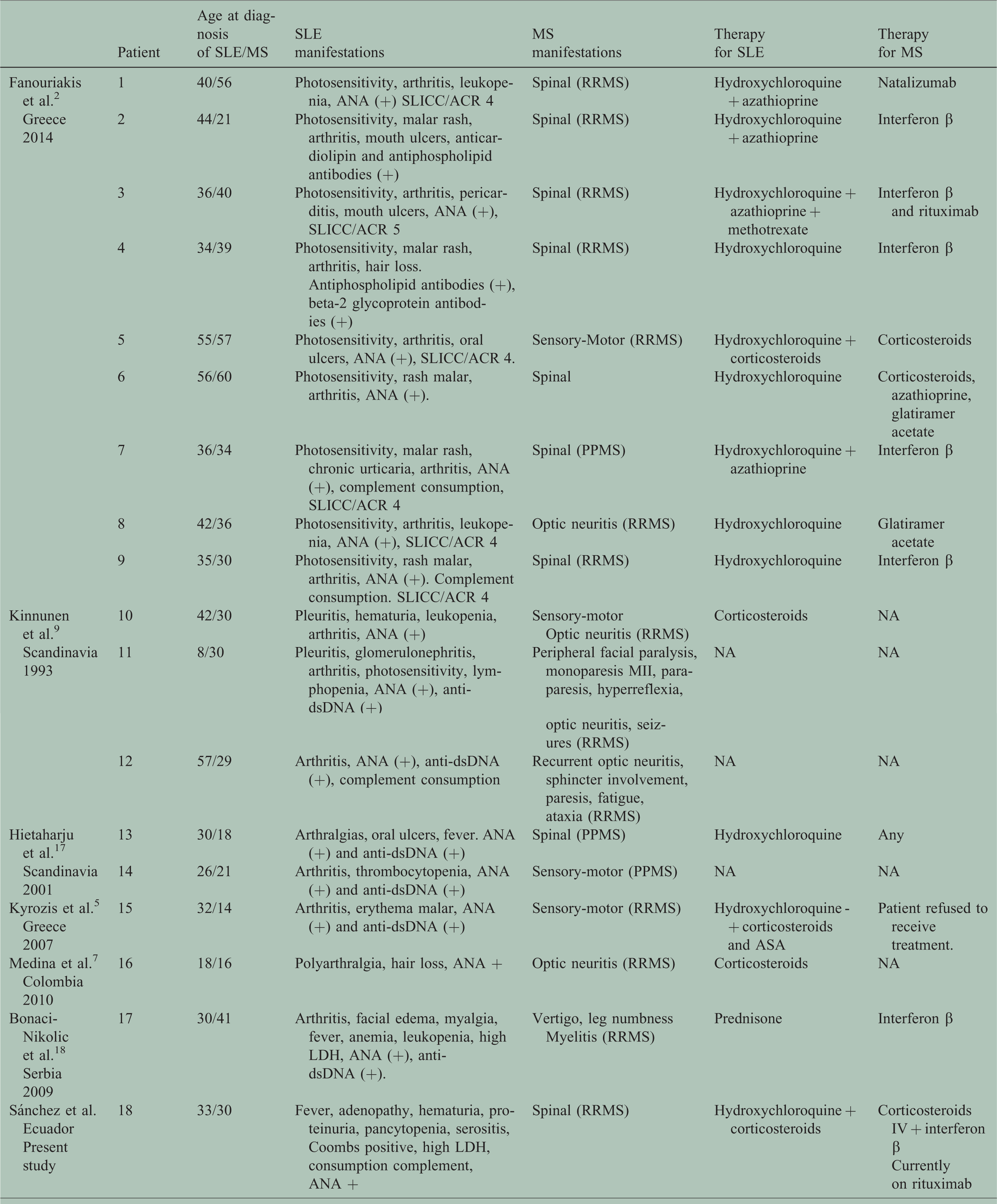
SLE: systemic lupus erythematosus; IV: intravenous; MS: multiple sclerosis; RRMS: relapsing–remitting multiple sclerosis; PPMS: primary progressive multiple sclerosis; ANA: antinuclear antibodies; SLICC/ACR: Systemic Lupus International Collaborating Clinics/American College of Rheumatology; NA: not applicable; LDH: lactate dehydrogenase; anti-dsDNA: anti-double-stranded DNA.
Our patient received subcutaneous INF beta-1a three times a week; this treatment was chosen because INF beta-1a has demonstrated efficacy through phase III clinical trials, 19 and it was the only medication available in Ecuador for the treatment of RRMS. Regarding the IFNs, in patients with SLE, type I INFs have been shown to promote the activation of the immune system and alter regulatory mechanisms, contributing to inflammation and tissue damage. 20 Drug-induced SLE is defined as a lupus-like syndrome related to continuous drug exposure that resolves after discontinuation of the offending drug. 21 However, few case reports have shown the development of SLE in patients with MS treated with INF.22–24 This contrasts with what happened in our patient, since the symptoms of SLE were present when the medication was withdrawn and worsened despite receiving treatment with hydroxychloroquine. We believe that previous infection with the dengue virus could have triggered the expression of type I INF and the subsequent development of SLE as demonstrated in studies in which SLE developed in individuals who were exposed to live virus vaccines.20,22,23 Additionally, IFN beta has been shown to induce the death of podocytes and prevents their differentiation from their precursors, making this treatment a contraindication for patients with lupus nephritis. 20
At present, there are very few therapies available for the concomitant treatment of SLE and MS. Management of SLE often depends on disease severity and disease manifestations (CNS involvement and diffuse proliferative renal disease). Hydroxychloroquine together with nonsteroidal anti-inflammatory drugs and analgesics are recommended in SLE with mild activity; prednisone together with methotrexate, azathioprine or mycophenolate mofetil (MMF) are recommended in SLE with moderate activity; and, in patients with severe activity but without renal damage or CNS involvement, cyclophosphamide, leflunamide or the combination of prednisone with MMF or rituximab are recommended. 25 In class III SLE glomerulonephritis, as in the case of our patient, an induction therapy based on methylprednisolone is required together with cyclophosphamide or MMF followed by maintenance therapy based on MMF, azathioprine or cyclophosphamide in low doses. 26
Rituximab is recommended in SLE with severe neurological, hematological, or renal damage that does not respond to first-line treatments. A study has shown that rituximab can be an effective and well-tolerated therapeutic option for refractory lupus nephritis.26–28 In MS, the immunosuppressants MMF, azathioprine, methotrexate and cyclophosphamide have been studied; however, their efficacy is not yet well established. A retrospective study has shown that 55% of patients had no evidence of disease activity when followed up with cyclophosphamide as induction therapy. 29 Another retrospective study showed that MMF reduced annualized relapse rate and EDSS remained stable between initiation and one year after the beginning of MMF. 30 A multicenter, randomized, non-inferiority trial has shown that efficacy with azathioprine was not inferior to that of IFN beta for patients with RRMS. 31 However, it is necessary that the efficacy of these drugs be demonstrated in phase III clinical trials and, if possible, be compared with disease-modifying therapies (DMTs).
Adrenocorticotropic (ACTH) hormone gel was approved by the United States Food and Drug Administration as a treatment for relapsing MS in 1978 and a treatment option for SLE in 1952.32,33 ACTH has anti-inflammatory and immunomodulatory effects due to activation of central and peripheral melanocortin receptors. 34 In MS, a systematic review demonstrated that ACTH or corticosteroids were effective over the short term in improving symptoms, thus favoring recovery. 35 With regard to SLE patients with moderate or severe active SLE, an open-label study showed that ACTH gel may provide significant disease activity reduction. 33 Another retrospective study has shown that ACTH appears to be safe and well tolerated after six months of SLE treatment with significant reduction of disease activity. 36
Our patient received treatment with rituximab, the efficacy of which in MS has been shown in observational and phase II studies. Hauser et al. have shown that compared with placebo, rituximab reduced inflammatory brain lesions and clinical relapses for 48 weeks. 37 Spelman et al. have shown that rituximab was superior to first-generation DMTs with respect to relapse control and tolerability. 22 An observational study showed that the rate of clinical relapses or neuroradiologic disease activity was significantly lower for rituximab when compared with injectable DMTs and dimethyl fumarate, with a tendency for a lower rate of relapses; this seems to also be the case when compared with natalizumab and fingolimod. 38 Our patient had stable RRMS and she received IFN before switching to rituximab. On this point, an open-label phase II multicenter study showed that in patients with stable RRMS, a treatment switch from INF or glatiramer acetate to rituximab was associated with reduction of disease activity measured by MRI and levels of CSF neurofilament light chain. 39 Also, rituximab seems to have improved effectiveness and tolerability compared with fingolimod in stable RRMS patients who switch from natalizumab because of JC virus antibody positivity. 40 Finally, an observational study has shown that rituximab was safe and effective in patients with RRMS who failed to respond to first- and second-line therapies and also a useful option for patients with concomitant autoimmune disorders such as in our case report. 41
In conclusion, the distinction between MS and SLE is a diagnostic challenge for the neurologist, and the presence of both diseases should be considered in patients with clinical neurologic manifestations of MS who present with typical systemic manifestations of SLE.
Footnotes
Conflicts of interest
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.