
Abstract
Background
Myelin oligodendrocyte glycoprotein (MOG) is a candidate primary target of the autoimmune attack on the central nervous system (CNS) in multiple sclerosis (MS). However, the physiological function of MOG has been unclear for a long time.
Objective
We propose that MOG has a central role in the regulation of tolerance and autoimmunity.
Conclusion
The interaction of MOG with DC-SIGN, an innate antigen receptor of myeloid antigen-presenting cells (m-APCs), present inside the CNS (microglia) or in draining lymph nodes (dendritic cells; DCs), keeps these cells in an immature/tolerogenic state. We postulate that this tolerogenic mechanism may be disturbed in MS by unknown factors.
Introduction
It has been proposed that autoimmunity in multiple sclerosis (MS) is not caused by an exogenous factor such as infection (outside-in), but by a dysregulated immune reaction against damage within the central nervous system (CNS; inside-out paradigm). 1 The cause of the injury is not known, but could be instability of myelin-axon units, resulting in impaired trophic support to axons by astrocyte-oligodendrocyte-myelin sheath complexes. 2 However, myelin degeneration per se, such as associated with trauma or ageing or in animal models, does not inevitably evoke autoaggressive immunity, although this can be observed when injury and concomitant glia activation accumulate. Despite the massive release of multiple myelin as well as non-myelin antigens after traumatic brain injury, a narrow B cell response against astrocytic glial fibrillary acidic protein (GFAP) can be detected, but not against myelin. 3 This raises the question why autoimmunity against myelin is rare in these disorders, but commonly observed in MS.
Here we propose the viewpoint that this can be explained by the key homeostatic task of the CNS myelin constituent myelin oligodendrocyte glycoprotein (MOG), being to keep professional antigen-presenting cells (APCs) present in the CNS (microglia) of draining lymph nodes (dendritic cells; DCs) in an immature/tolerogenic state. This homeostatic function depends on the glycosylation of MOG 4 and may be disturbed by genetic or presently unknown glia-toxic factors, which have been found in MS patients. 5
Regulation of T cell autoimmunity by myeloid antigen presenting cells (m-APCs)
Professional APCs ingest and degrade protein antigens within endolysosomal compartments, and present degradation products (peptides) via major histocompatibility complex (MHC) class II molecules to CD4+ T cells.6,7 APCs of the myeloid lineage (m-APC), comprising DCs and macrophages (MΦ), and of the lymphoid lineage (B cells) are discerned. M-APCs, DCs in particular, have a key regulatory role in T cell tolerance and immunity against self-antigens. 8 M-APCs are located in peripheral tissues (DCs), within the CNS (microglia) and in the cervical and lumbar lymph nodes (DCs) that drain the brain and spinal cord. 9 The m-APCs continuously collect antigens and cell debris from the environment and are informed via innate pathogen recognition receptors (PRRs) about the actions that need to be taken. Examples of PRRs are the Toll-like receptors (TLRs), which in general (albeit not all) relay activation signals, 10 and the C-type lectin receptors (CLRs), which in general (albeit not all) relay inhibitory signals.4,11 We proposed that by integrating the strength of inhibitory and activation signals the m-APC ‘learns’ whether an immature/tolerogenic state should be maintained or whether differentiation to a mature/immunogenic state is required. 12
The Yin-Yang paradigm proposed that under homeostatic conditions (scenario 1) input signals received via CLR counterbalance those relayed through TLRs. In the healthy tissue TLRs activating danger signals from pathogens (pathogen-associated molecular patterns (PAMPS)) or cell damage (damage-associated molecular patterns (DAMPS)) are low or absent, while CLR binding glycosylated antigens are abundant. Conceptually, antigen presentation by immature m-APCs induces regulatory T (Treg) cells (Figure 1). In the case of threat by infection or tissue injury (scenario 2), PAMPS and DAMPS are released and bind to TLRs, thus increasing the strength of activation signals that induce DC maturation. In the mature/immunogenic state, m-APCs express co-stimulatory molecules for full activation of T cells together with cytokines (interleukin (IL)-12, -23) that determine their pro-inflammatory function. When the pathogen has been cleared, the CLR/TLR balance and homeostasis are restored and remission is induced. We predicted that clinical conditions affecting the normal glycosylation of tissue antigens (scenario 3) can cause permanent instability of the CLR/TLR equilibrium, which potentially results in chronic T cell activation without remission.
Yin-Yang concept of autoimmunity: The type of autoimmune reaction that DCs induce when presenting antigen to T cells depends on environmental signals they receive through innate pattern recognition receptors. In the homeostatic situation (scenario 1; no disease) antigen is sampled from the environment in the absence of danger; i.e. inhibitory signals received via CLRs are in balance with activating signals via TLRs. The APCs remain in an immature/tolerogenic state that induces Treg → no disease (b). When antigen is recognised in the context of danger signals relayed through TLRs (scenario 2; transient autoimmunity), the DCs mature to an immunogenic state that activates autoaggressive T cells. When the danger has been cleared homeostasis is restored; newly formed Treg cells dampen autoimmune process. When scenario 2 occurs in individuals with a glycosylation defect, restoration of homeostasis and disease remission cannot occur (scenario 3; chronic autoimmunity). DCs: dendritic cells; CLRs: C-type lectin receptors; TLRs: Toll-like receptors; APCs: antigen-presenting cells; Treg: regulatory T cells.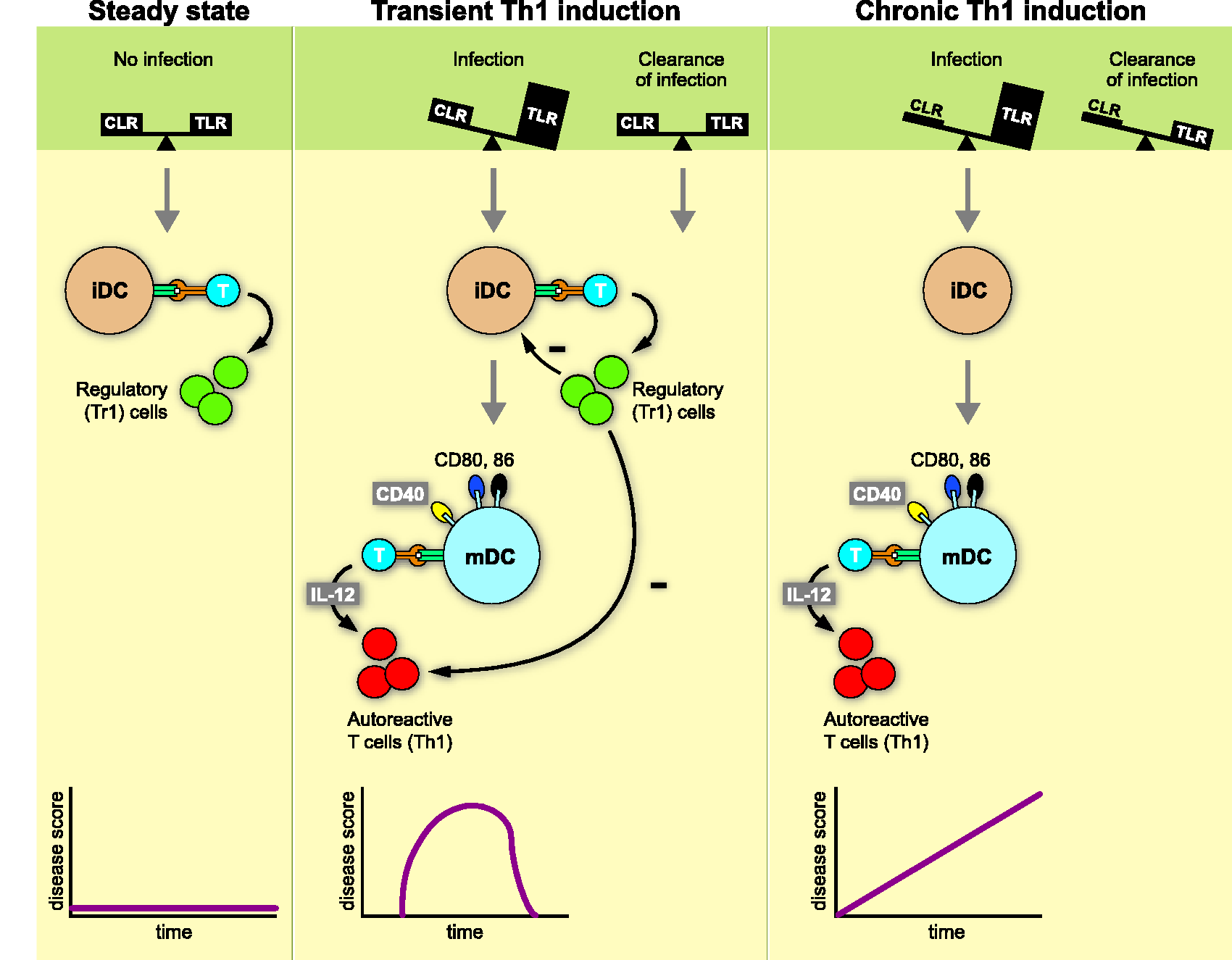
MOG as a dominant tolerogen for CNS myelin
MOG is only expressed by oligodendrocytes within the CNS of mammalian species. The MOG gene is highly conserved among species and the molecule had thus far no defined physiological function. MOG-deficient mice develop normally without obvious abnormalities in the myelination of their CNS axons. 13 For a long time, MOG was known only as a primary target of demyelination-inducing autoantibodies. 7 It is difficult to understand, however, that without a beneficial physiological function the prominent expression of MOG within a vulnerable organ such as the brain would have survived evolutionary selection. Such an important beneficial role was recently found. 4
MOG is produced by healthy human oligodendrocytes as an immunoglobulin (Ig)-like molecule decorated with a fucosylated N-linked glycan attached to the asparagine residue at position 31 within the extracellular domain. The glycan epitope supports recognition by the CLR DC-SIGN, which is expressed on microglia and DCs. As DC-SIGN does not bind to MOG-deficient myelin, MOG is the only identified ligand of DC-SIGN in CNS myelin. MOG exerts a potent regulatory function. Stimulation of DCs with the TLR4 ligand bacterial lipopolysaccharide (LPS) in the presence of myelin particles binding via MOG to DC-SIGN induced an anti-inflammatory state, characterised by predominant production of IL-10 and suppressed inflammasome formation. 4 Intriguingly, co-incubation of DCs with LPS and de-fucosylated myelin, which abrogates DC-SIGN binding, evoked inflammasome activation and induction of pro-inflammatory T cells. 4 These data indicate a crucial physiological function of MOG, namely prevention of T cell autoimmunity by dampening m-APC maturation. In line with this concept, it was found that mice with defects in the N-linked glycosylation of proteins spontaneously develop MS-like pathology and disease. 14
MOG as a dominant autoimmunogen
The presence of MOG in myelin is essential for chronic experimental autoimmune encephalomyelitis (EAE) development in mice, 15 rats 16 and non-human primates. 17 Taking advantage of the phylogenetic proximity to humans, marmoset monkeys are frequently used for proof-of-principle experiments before clinical trials are initiated in humans and also to explore hypotheses regarding the putative pathophysiological events leading to MS. EAE development in marmosets immunised with human MOG as a non-glycosylated recombinant protein in the presence of danger signals (from heat-killed mycobacteria in complete Freund’s adjuvant; FA = IFA) involves two distinct autoimmune pathways.18,19 One pathway leads to brain and spinal cord white matter inflammation via MHC class II/Caja-DRB*W1201-restricted CD4+ T helper 1 cells specific for the epitope MOG24-36. The second pathway leads to demyelination of brain and spinal cord white and grey matter via MHC class I/Caja-E-restricted CD8+ effector memory cytotoxic T lymphocytes (CTLs) specific for the epitope MOG40-48. Anti-MOG B cells have different roles in these two pathways. In the first pathway B cells are needed for the production of anti-MOG antibodies, which cause focal demyelination via mobilisation of complement and/or MΦ. 20 The second pathway operates independently of autoantibodies; here B cells are the requisite APCs of demyelinating CTLs, provided that they are infected with a γ1-herpesvirus, such as human Epstein-Barr virus (EBV) or marmoset CalHV3. 21 This recently discovered pathway has not been found in current rodent EAE models.
The picture changed when marmosets were immunised with rhMOG formulated with incomplete FA (IFA), which lacks detectable danger signals. In this highly refined EAE model encephalitogenic CD4+ T cells against MOG24-36 were activated and antibodies binding CNS myelin were induced, but T cell reactivity against the CTL epitope MOG40-48 was undetectable. 22 Nevertheless, when marmosets were immunised with a mixture of synthetic MOG14-36 and MOG34-56 peptides in IFA, activation of encephalitogenic T cells against the MOG40-48 epitope could be detected. This finding indicates that MOG24-36-specific CD4+ Treg cells are present in the natural repertoire and that, when activated by the MOG14-36 peptide, they can prevent activation of CD8+ CTL cells against the flanking epitope MOG40-48. This phenomenon is known as linked-suppression.
Accumulating evidence demonstrates that the non-responsiveness of marmosets to the MOG40-48 epitope is broken when the MOG40-48 epitope is presented by B cells infected with the EBV-like lymphocryptovirus (LCV) CalHV3. This is the case in the model induced with MOG34-56 peptide in IFA. 23 Recent work shows that LCV infection skews the APC function of B cells from anti-inflammatory/tolerogenic to pro-inflammatory/immunogenic by altering the processing of the MOG34-56 peptide (Jagessar et al., manuscript in preparation).
Conclusion
We propose that the explanation for the absence of anti-myelin autoimmunity in trauma and the presence in MS may be in the inflammatory condition of the lesion. Oligodendrocytes produce differently glycosylated MOG under inflammatory compared to homeostatic conditions. Alteration of the normal glycosylation impairs the capacity of MOG to interact with DC-SIGN and to keep APCs, present inside the CNS (microglia) or in DCs, in an immature/tolerogenic state. Due to the altered glycosylation of MOG, activation of m-APC in CNS is induced, leading to amplification of local presentation of myelin antigens, (re-) activation of T cells and influx of additional cell types and soluble factors into the CNS, which together induce full-blown MS lesion formation.
Footnotes
Acknowledgement
The authors thank Mr Henk van Westbroek for the artwork.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest
None declared.