
Abstract
Background
Ofatumumab is approved for treating relapsing multiple sclerosis (RMS). Examining tolerability will enable understanding of its risk–benefit profile.
Objective
Report the tolerability profile of ofatumumab in RMS during treatment of up to 4 years and the effect of pre-medication.
Methods
Cumulative data from the overall safety population included patients taking continuous ofatumumab or being newly switched from teriflunomide. Injection-related reactions (IRRs) by incidence and severity, and post-marketing surveillance data, with an exposure of 18,530 patient-years, were analyzed.
Results
Systemic IRRs affected 24.7% of patients (487/1969) in the overall safety population; most (99.2% [483/487]) were mild (333/487) to moderate (150/487) in Common Terminology Criteria for Adverse Events severity; most systemic IRRs occurred after first injection. Local-site IRRs affected 11.8% (233/1969) and most (99.6% [232/233]) were mild/moderate. Incidence and severity of systemic and localized IRRs were similar between continuous and newly switched patients across repeated injections. Systemic IRR incidence and severity were not substantially affected by steroidal or non-steroidal pre-medication. Post-marketing surveillance identified no new tolerability issues.
Conclusion
Ofatumumab is well tolerated, displays a consistent safety profile during continuous use or after switching from teriflunomide and does not require pre-medication. This enables home management of RMS with a high-efficacy treatment.
Keywords
Introduction
Multiple sclerosis (MS) is a chronic, autoimmune demyelinating disease of the central nervous system. 1 Available disease-modifying treatments (DMTs) typically focus on reducing relapses, limiting disease worsening and maintaining health-related quality of life. 2 Advances in the development of DMTs for relapsing forms of MS enabled additional factors, including route and frequency of administration, and requirements for concomitant pre-medication prior to administration, to be considered when selecting the most appropriate treatment. 1 Two key aspects involved in DMT decisions include the comparative efficacy and associated safety profile of each therapy. 2 The safety profile can also adversely affect the tolerability of, and adherence to, DMTs when used long term. 3 A previous systematic assessment of DMTs widely used in the treatment of MS found that the most common tolerability issues for legacy injectables were flu-like symptoms and injection-site reactions, or gastrointestinal symptoms for some oral therapies.3,4
Ofatumumab is the first fully human anti-CD20 monoclonal antibody approved for the treatment of relapsing MS (RMS).5,6 Ofatumumab induces B-cell depletion through binding to two regions on a unique, conformational epitope of the CD20 receptor.7–9 This region includes the large and small extracellular loops, leading to strong binding affinity and a slow off-rate. 10 Ofatumumab induces efficient B-cell depletion through complement pathway-dependent cytotoxicity and antibody-dependent cell-mediated toxicity.8,9,11–13 Pharmacokinetic studies and the clinical development programme have determined that the optimum dosing of subcutaneous (SC) ofatumumab for B-cell depletion is 20 mg every 4 weeks, following loading doses given at weeks 0, 1, 2 and 4.8,12 The pivotal ASCLEPIOS I and II teriflunomide-controlled phase 3 trials, lasting up to 30 months, met their primary efficacy endpoint of adjusted annualized relapse rate (ARR) being significantly lower in the ofatumumab group than the teriflunomide arm (ASCLEPIOS I ARR: ofatumumab = 0.11, teriflunomide = 0.22, p < 0.001; ASCLEPIOS II ARR: ofatumumab = 0.1, teriflunomide = 0.25, p < 0.001). 9 Thus far, ofatumumab has shown a favourable safety profile with no new safety risks being identified in open-label extension (OLE) studies.7–9,11
Injection-related reactions (IRRs) are the most frequently reported adverse event (AE) in the ofatumumab clinical development programmes,7–9,14,15 highlighting the need to further investigate its tolerability. Phase 2 trials suggested that most IRRs were mild to moderate in severity, primarily occurring after the first ofatumumab dose.14,15 In APLIOS, which determined the bioequivalence of ofatumumab between prefilled syringe and autoinjector administration methods, IRRs affected 25.0% of patients after first injection, 8.1% after the second and less than 2.8% in subsequent doses. 8 The most common systemic IRRs were headache (12.7%), chills (8.8%) and fever (8.5%), with the majority resolving within 48 h. 8 In the phase 3 ASCLEPIOS I and II trials, IRRs, defined as occurring within the first 24 h after injection, affected 20.2% of the ofatumumab group, compared with 15.0% receiving teriflunomide with a placebo injection.9,11 Across these two pivotal trials, IRRs were most frequent after the first ofatumumab dose; they were predominantly mild to moderate and most resolved without medical intervention.7–9,11 A trial investigating intravenous (IV) ofatumumab demonstrated a higher incidence of systemic IRRs 16 than SC administration.7–9,11 Long-term investigation of the tolerability profile and management of IRRs in SC ofatumumab treatment would further inform developments in clinical care.
The ongoing OLE study of ofatumumab, ALITHIOS, includes patients from the phase 2 APLIOS and APOLITOS studies, along with ASCLEPIOS I and II. Herein, we utilize data from the overall safety population and post-marketing surveillance to conduct an analysis of ofatumumab's tolerability profile, to expand on existing data from pivotal trials and inform the clinical community better.
Materials and methods
Clinical development programme and OLE trial designs and patients
Following the four trials in the sponsor's (Novartis Pharma AG) clinical programme investigating ofatumumab (ASCLEPIOS I and II [NCT02792218 and NCT02792231], 9 APLIOS [NCT03560739] 8 and APOLITOS [NCT03249714] 7 ), patients could choose to enter the OLE ALITHIOS (NCT03650114).
The trial design, inclusion and exclusion criteria, description of the safety follow-up period, patient demographics and RMS characteristics for ASCLEPIOS I and II, APLIOS and APOLITOS have been described previously.7–9 The OLE ALITHIOS started in December 2018. The tolerability data from ALITHIOS included in this manuscript are derived from a data cut-off of 25 September 2021. For patients randomized to ofatumumab in ASCLEPIOS I and II, this is a cumulative ofatumumab exposure of up to 4 years with a median exposure of 35.8 months.
ALITHIOS trial oversights: Standard protocol approvals, registrations and patient consent
ALITHIOS was designed by the sponsor Novartis and is being conducted in accordance with the International Conference on Harmonisation guidelines for Good Clinical Practice, local regulatory requirements and the principles of the Declaration of Helsinki.17,18 The trial protocol was approved by an independent ethics committee or institutional review board for each trial site, and all patients provided written informed consent before starting trial-related procedures.
IRR analysis populations in the ofatumumab clinical development programme
The overall safety population for this analysis includes data from 1969 patients with MS, summarized in Figure 1. Patients treated with ofatumumab in ASCLEPIOS I and II, APLIOS or APOLITOS, regardless of whether they enrolled in the OLE ALITHIOS, comprise the continuous ofatumumab group. Patients randomized to teriflunomide in ASCLEPIOS I and II and switching to ofatumumab upon enrolment in ALITHIOS comprise the newly switched group.
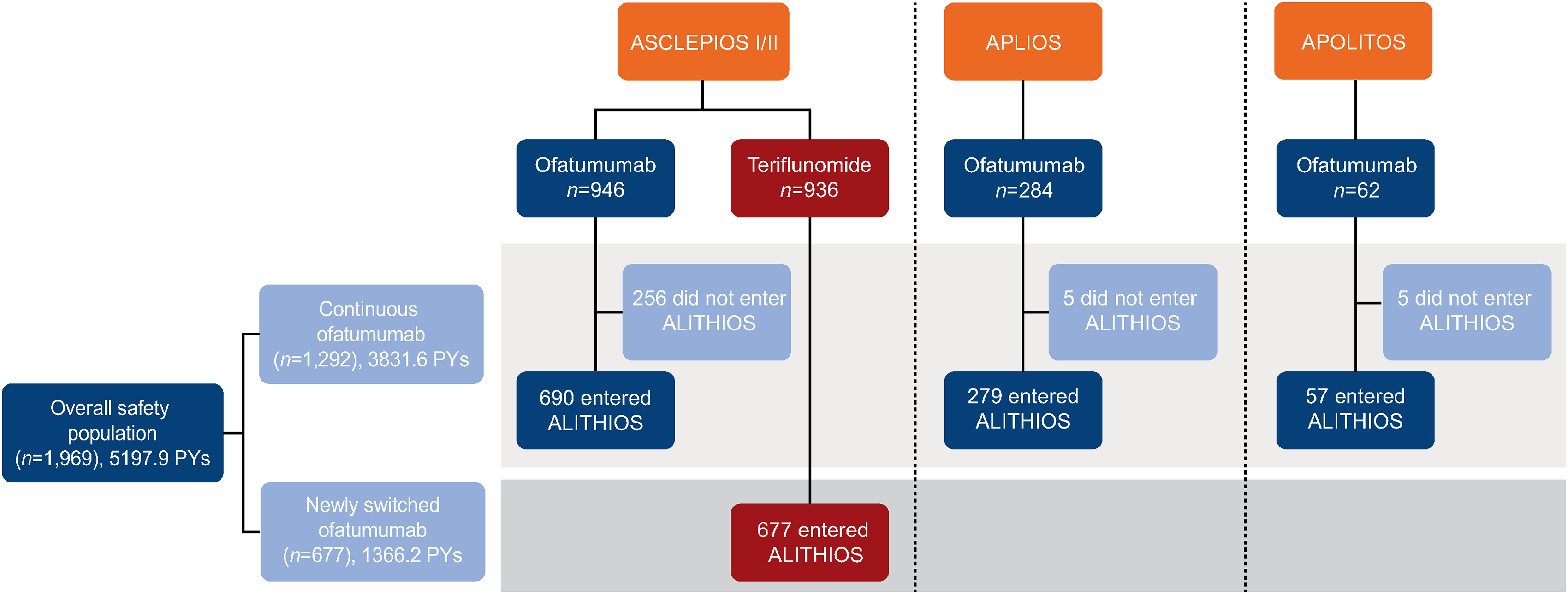
Patient disposition of safety analysis set. Patients were either randomized to or switched to ofatumumab during the core study. PY: patient-year.
Trial IRR data collection
In the trial population, investigators reported IRRs as either systemic or local-site IRRs. By definition, systemic IRRs occurred within 24 h after administration of the injection and were assumed to be injection-related. Local-site IRRs around the injection site could be reported without time limit. IRR severity was recorded using the Common Terminology Criteria for Adverse Events (CTCAE) grading and categorized as: mild (grade 1), moderate (grade 2), severe (grade 3) or life threatening (grade 4). 19
The proportions of patients with systemic and local-site IRRs were analysed by treatment group, against the injection sequence number (injections 1–10), as well as cumulatively across all injections. At the discretion of trial investigators, patients were prescribed pre-medication prior to ofatumumab administration, in the form of steroids (methylprednisolone 100 mg or equivalent), acetaminophen or anti-histamines (over-the-counter medications). Pre-medication was not mandatory, and some patients were not prescribed any. The incidences of IRRs by pre-medication category, severity, symptoms and injection sequence number were also assessed. Patients received the first four injections in a clinical setting under medical supervision; following this, most patients self-administered ofatumumab at home.
Post-marketing surveillance for IRRs
The post-marketing data cover the period from ofatumumab US Food and Drug Administration approval for RMS, 20 August 2020, to a cut-off on 25 March 2022. In the post-marketing setting, reports are often non-specific or incomplete. As such, a broad search strategy was used, and further evaluation was performed to identify true cases of systemic IRRs. The post-marketing data were taken from a global cohort and identified using Medical Dictionary for Regulatory Activities search terms. Cases identified were specific IRRs immediately post-injection, or presenting a plausible temporal association within 24 h. Cases likely due to confounding factors, such as infection, RMS relapse or concurrent illness, were excluded. The medical review contained all fatal and life-threatening events received from both healthcare professionals (HCPs) and non-HCPs, including patients and caregivers, along with other medically confirmed serious injection reactions. Cytokine release syndrome and hypersensitivity are presented as a standalone topic, to differentiate potential cases from systemic or localized IRRs.
Results
Summary of patient demographics
Baseline patient demographics and disease characteristics have been previously reported for ASCLEPIOS I and II, 9 APLIOS 8 and APOLITOS. 7 The baseline demographics for the 1969 patients in the safety analysis population are described in Table 1. In the overall safety population, 86.5% of patients (n = 1703) were enrolled in the OLE ALITHIOS. 11 Across the safety population, the mean (standard deviation [SD]) age of patients was 38.7 (9.2) years, and 68.3% of patients (n = 1345) were female. Mean (SD) time since diagnosis was 6.4 (6.2) years and since symptom onset was 9.0 (7.3) years, while mean (SD) Expanded Disability Status Scale score was 2.9 (1.4).
Demographics and disease characteristics (safety analysis set).
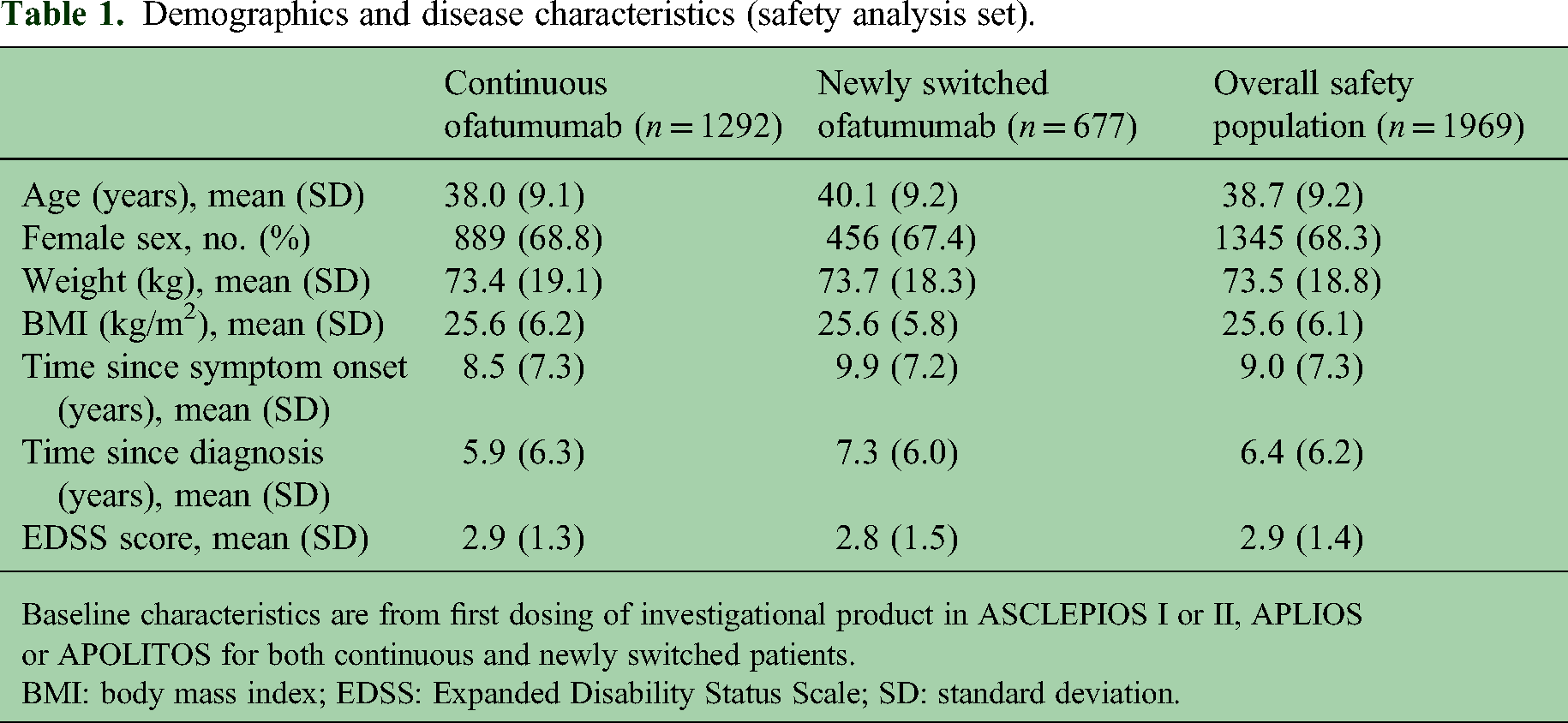
Baseline characteristics are from first dosing of investigational product in ASCLEPIOS I or II, APLIOS or APOLITOS for both continuous and newly switched patients.
BMI: body mass index; EDSS: Expanded Disability Status Scale; SD: standard deviation.
IRR analysis populations in ofatumumab clinical development programme
The overall safety population comprised 1292 patients in the continuous ofatumumab group and 677 patients in the newly switched ofatumumab group. The patient populations, preceding trial and ofatumumab treatment history are summarized in Figure 1. The total safety population incorporated data from 5197.9 patient-years (PY): 3831.6 PY from the continuous ofatumumab and 1366.2 PY from the newly switched ofatumumab groups.
Compliance was high, with 94.9% of patients (n = 1868) compliant with therapy in the overall safety population, at a cut-off of 25 September 2021. The compliance was similar between the continuous (95.1% [n = 1229]) and newly switched (94.4% [n = 639]) groups.
Summary of systemic IRRs incidence and severity
Across the safety population, the incidence of at least one systemic IRR across any injection was observed in 24.7% of patients (n = 487) and was similar between the continuous and newly switched ofatumumab groups, at 25.7% (n = 332) and 22.9% (n = 155), respectively. Systemic IRRs were highest with the first ofatumumab administration, with a similar incidence occurring in the continuous ofatumumab (17.0% [n = 219]) and newly switched ofatumumab groups (18.3% [n = 124]). The incidence decreased substantially with subsequent injections, as shown in Figure 2. Nearly all systemic IRRs were CTCAE mild to moderate in severity (99.2% [483/487]) and non-serious (99.4% [484/487]). There were 0.3% of patients (5/1969) with systemic IRRs who discontinued ofatumumab treatment; 4/677 (0.6%) in the newly switched group with mild or moderate systemic IRRs and 1/1292 (0.1%) in the continuous group with a severe IRR. There were no life-threatening IRRs or instances of cytokine release syndrome reported. Approximately 8.5% of participants (166/1963) experienced more than one systemic IRR, as summarized in Table 2.
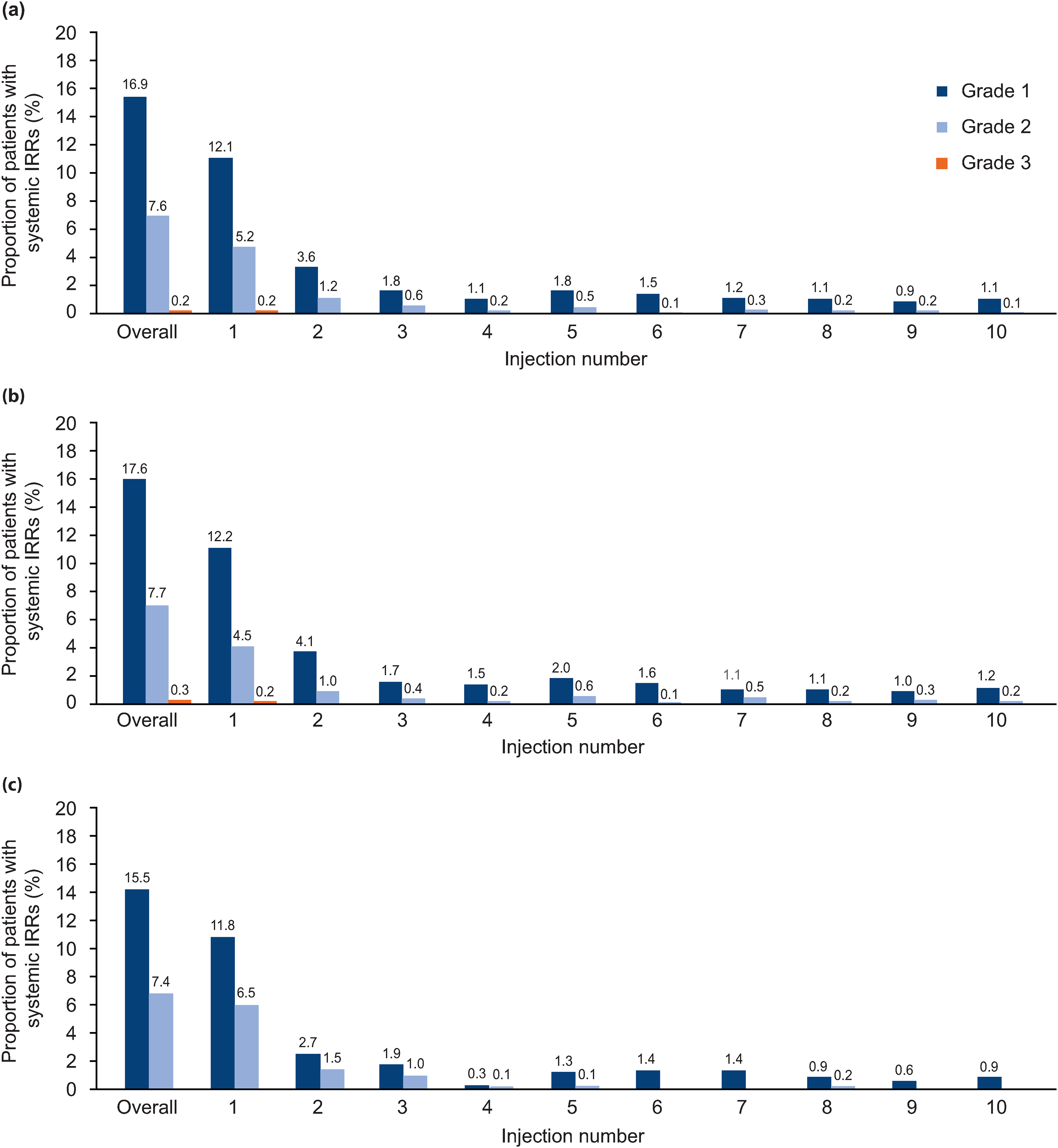
Incidence of systemic IRRs by injection and severity in (a) the overall safety population, (b) the continuous ofatumumab group and (c) the newly switched ofatumumab group. IRR: injection-related reaction.
Number of participants experiencing more than one systemic or localized IRR.

All data are shown as % (n).
IRR: injection-related reaction.
The most common symptoms related to systemic IRRs in the overall safety population were fever (10.1% [n = 199]), headache (8.1% [n = 160]) and chills (6.5% [n = 128]), as summarized in Table 3. These were the three most common symptoms in both the continuous and newly switched groups.
Incidence of symptoms >5% in overall safety population related to systemic IRRs.
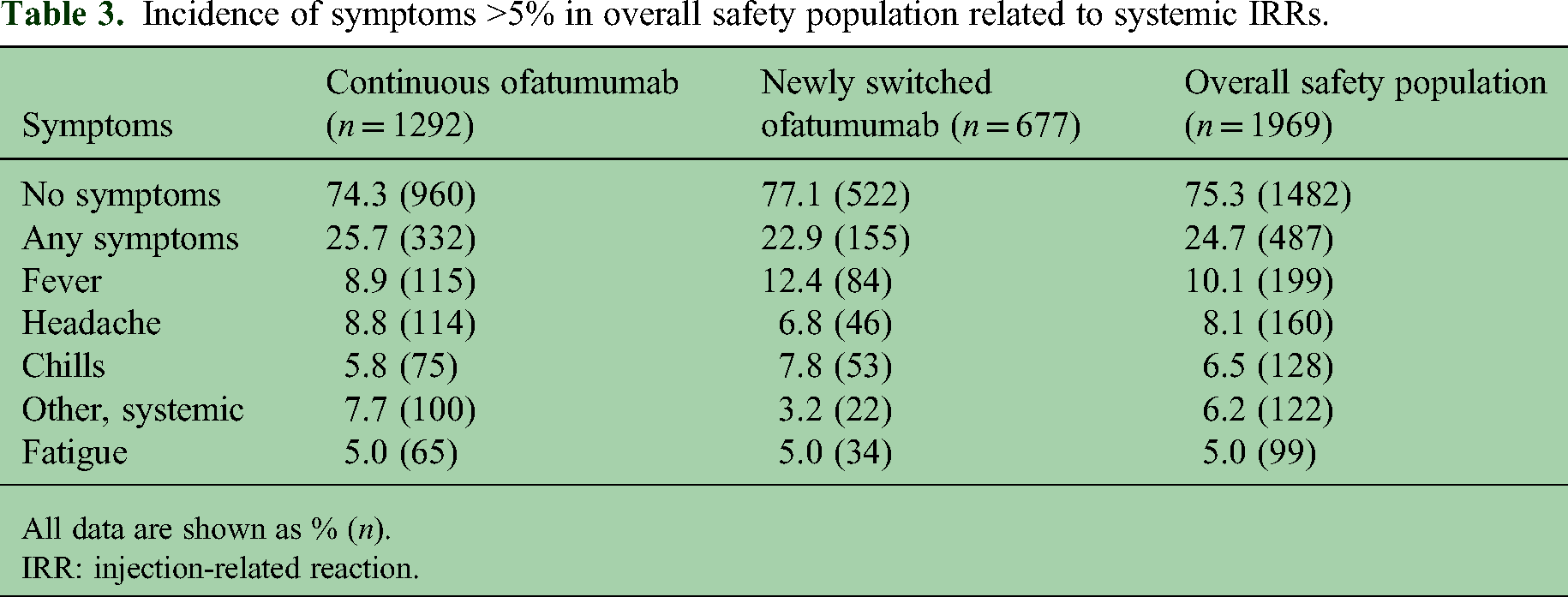
All data are shown as % (n).
IRR: injection-related reaction.
Summary of systemic IRR severity by pre-medication group
Prior to the first ofatumumab dose, 65.1% of patients (n = 1281) in the safety population received steroids, 9.6% (n = 190) received only non-steroidal pre-medication and 25.3% (n = 498) took no pre-medication. In general, the percentage of patients using pre-medication decreased by injection number, as summarized in Figure 3. Across the safety population, 74.7% of patients (1471/1969) took pre-medication for the first ofatumumab injection, 28.3% (555/1963) for the second and 23.8% (465/1956) for the third. The incidence of systemic IRRs by pre-medication category and injection number can be found in Supplementary Material; systemic IRRs were predominantly mild and occurring after the first injection. The severity of systemic IRRs experienced across all injections did not differ substantially by pre-medication group, or between continuous ofatumumab and newly switched ofatumumab patients, as shown in Figure 4.
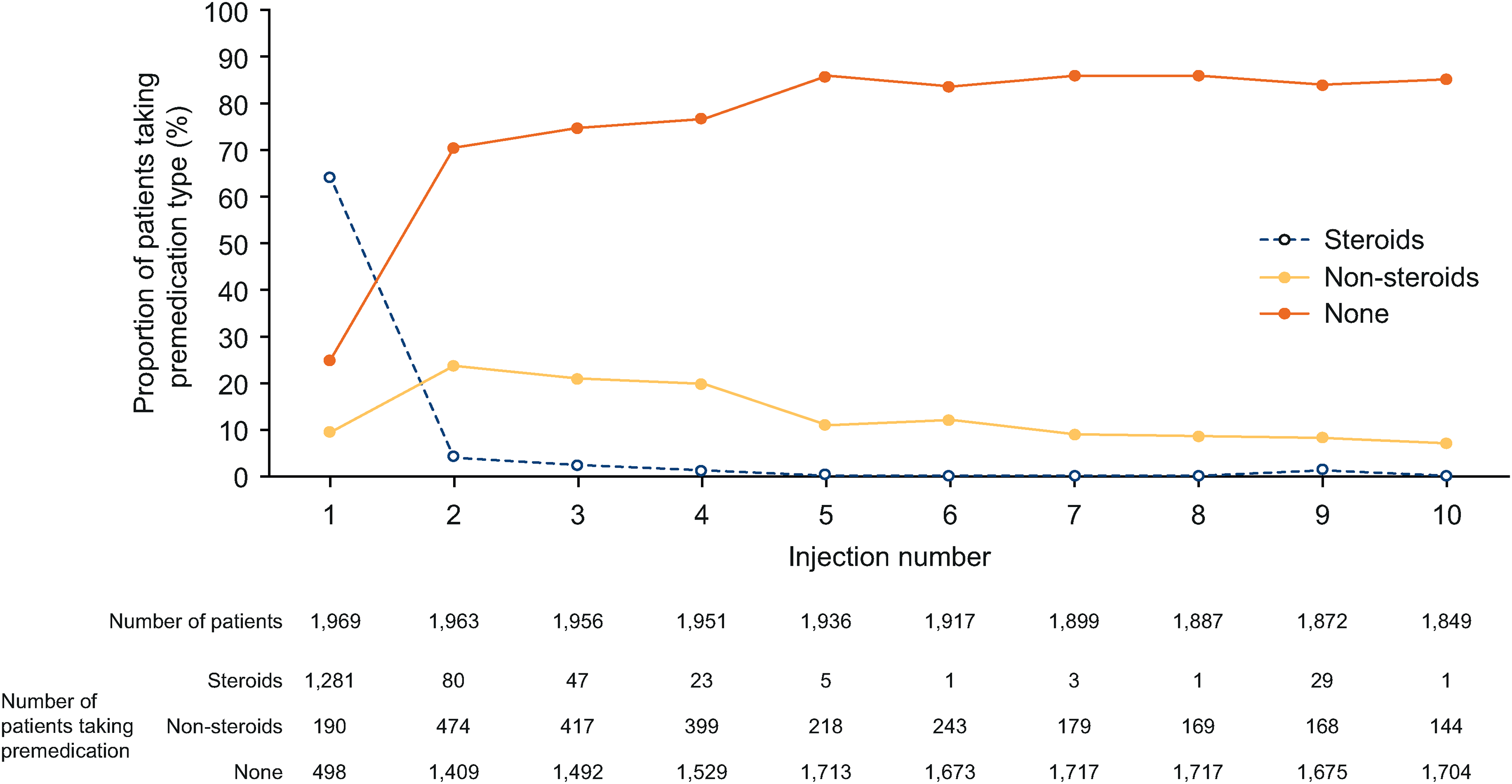
Proportion of pre-medication type administration by ofatumumab injection number.
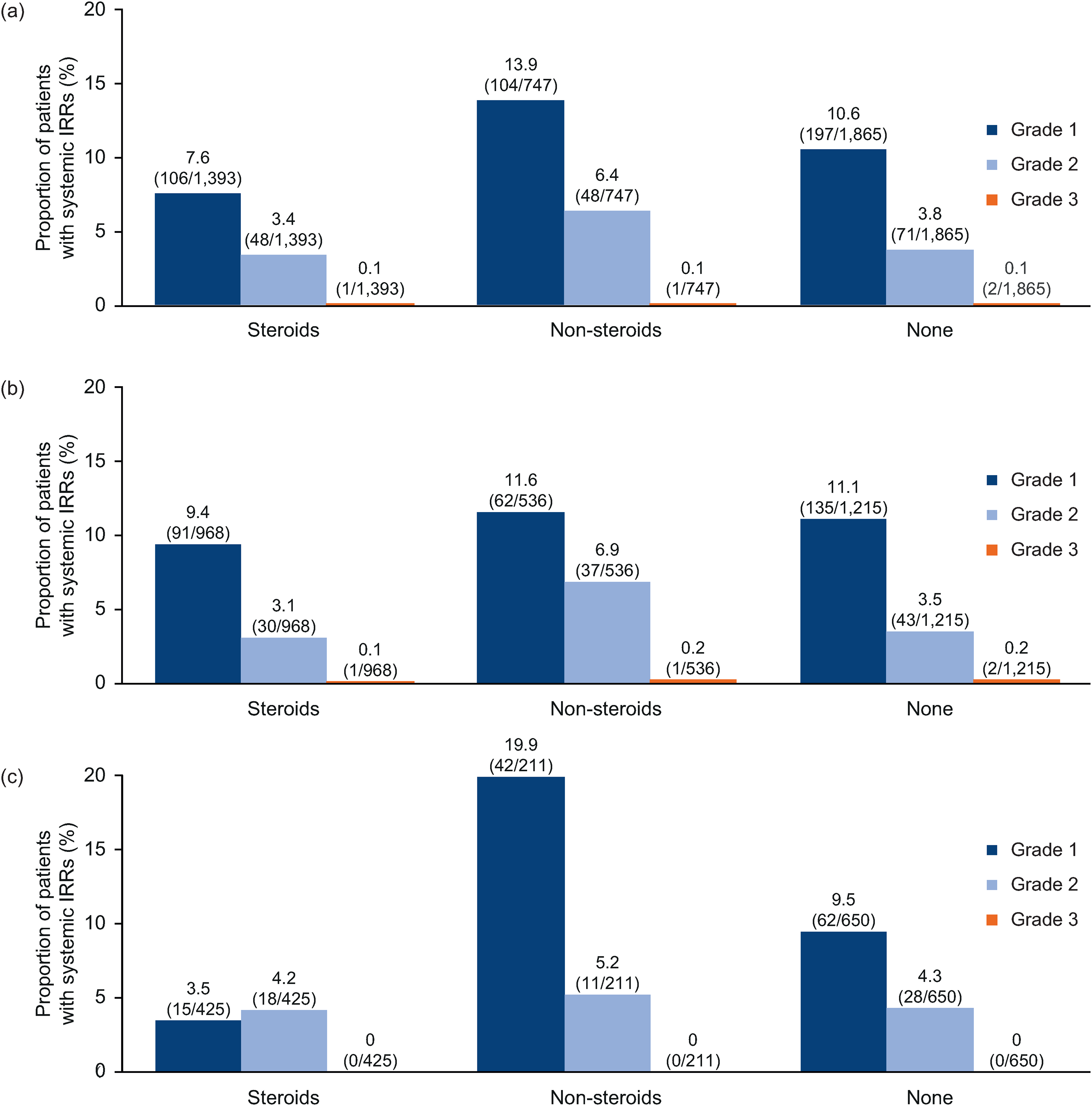
Severity of systemic IRRs by pre-medication in the (a) overall safety population, (b) continuous ofatumumab group and (c) newly switched ofatumumab group. The number of patients taking pre-medication is the number of patients taking pre-medication prior to at least one injection. (n/N) n = number of patients with systemic IRR, N = number of patients taking pre-medication type in population. IRR: injection-related reaction.
Most systemic IRRs were mild to moderate in CTCAE severity
Of the 1292 patients taking continuous ofatumumab, 74.9% (n = 968) took steroidal pre-medication prior to at least one ofatumumab injection, along with 62.8% of the newly switched group (n = 425). Furthermore, 41.5% of patients (n = 536) taking continuous ofatumumab took only non-steroidal pre-medication before at least one injection, as did 31.2% of the newly switched patients (n = 211). Between the continuous and newly switched groups and pre-medication types, systemic IRRs were broadly similar in incidence, with the majority reported as grade 1 or 2 in CTCAE severity.
Before at least one of the ofatumumab injections, 93.0% of patients (n = 1201) in the continuous ofatumumab group and 96.0% (n = 650) in the newly switched group did not premedicate. Systemic IRR incidence was similar between groups and most systemic IRRs were reported as grade 1 or 2 in severity.
Summary of local-site IRRs by treatment group
Overall, local-site IRRs affected 11.8% of patients (n = 233) in the safety population, with similar incidences in the continuous ofatumumab and newly switched groups, at 13.5% (n = 175) and 8.6% (n = 58), respectively. Local-site IRRs were typically mild, as shown in Figure 5.

Incidence of local-site IRRs by severity. IRR: injection-related reaction.
Local-site IRRs were nearly all mild to moderate (99.6% [232/233]) in severity and non-serious (99.6% [232/233]); one patient reported a grade 3 local-site IRR and none were life-threatening. One patient in the newly switched group discontinued treatment owing to grade 1 local-site IRRs of erythema, swelling and pain.
The most common local-site IRR symptoms were similar in frequency in the continuous and newly switched ofatumumab groups, as shown in Table 4. The three most common local-site IRRs in the overall safety population include erythema/redness affecting 6.7% (n = 132) and pain at the injection site affecting 3.9% (n = 76) of patients. Between the continuous and newly switched groups, the incidence of local-site IRR symptoms was similar. Approximately 6.1% of participants (120/1963) experienced more than one localized IRR, as summarized in Table 2.
Incidence of symptoms in >2% of treatment group related to local-site IRRs.

All data are shown as % (n).
IRR: injection-related reaction.
IRRs in post-marketing surveillance data
Post-marketing surveillance covered an estimated cumulative post-marketing exposure of 18,530 PY, until 25 March 2022. Cumulatively, there were 4855 cases of possible systemic IRRs; 178 of which were serious and 4677 were non-serious. Of the 178 serious cases, 39 were HCP confirmed. These cases were then further HCP-evaluated to confirm if they were true or potential systemic IRRs. Of these HCP-confirmed cases, 14 true or potential systemic IRRs were identified in total, 13 patients were hospitalized, and one did not require hospital care. Reported symptoms in more than two of these cases included pyrexia, chills and fatigue. There were no medically confirmed life-threatening or fatal systemic IRRs identified in the surveillance period. Potential local-site IRRs were reported in 566 patients; 99.3% (n = 562) were non-serious and the most common event was injection-site pain (n = 188). The review did not identify new safety risks from ofatumumab administration in a post-marketing setting.
Discussion
The overall safety population included data from 1969 patients with an exposure of 5197.9 PY. Findings suggest that most ofatumumab-associated IRRs are mild in nature and occurred after first injection, consistent with the clinical development programme and ongoing post-marketing surveillance.7–9 This analysis included patients taking continuous ofatumumab (median: 35.8 months) and patients newly switched from the teriflunomide group in ASCLEPIOS I/II upon entering ALITHIOS. Nearly all systemic IRRs were mild to moderate in severity, with no substantial difference exhibited between the continuous and newly switched ofatumumab groups. 4
Side effects and poor tolerability are primary reasons for discontinuation of DMTs in patients with RMS. 4 Gastrointestinal symptoms are notable with oral therapies, such as dimethyl fumarate, and flu-like symptoms are frequently experienced with injectables, such as interferon-beta, and lipoatrophy is frequently experienced with glatiramer acetate treatment.4,20,21 In clinical practice, 32–46% of patients discontinue oral or injectable DMTs within 5–31 months after initiation. 4 Conversely, in this tolerability analysis for ofatumumab, only 0.3% of patients with systemic IRRs in the overall safety population discontinued ofatumumab, suggesting a favourable and differentiated tolerability profile for ofatumumab.
Intravenously administered anti-CD20 B-cell-depleting therapies show varied tolerability profiles and require a higher dose that is administered in a clinical setting.8,22,23 Infusion-related AEs were reported in 78.3% of participants (54/69) within 24 h after rituximab administration. This contrasts with the lower incidences of ocrelizumab infusion-related reactions reported during the OPERA I and II trials (30.9% [126/408] and 37.6% [157/417], respectively) and the incidence of ofatumumab systemic IRRs during the ALITHIOS trial (24.8% [487/1969]). The majority of AEs being mild to moderate (CTCAE grade 1 or 2) in severity was consistent across all three anti-CD20 B-cell-depleting therapies (summarized in Table 5).11,22,24
Tolerability profile of selected anti-CD20 monoclonal antibody therapeutics for MS.

AE: adverse event; CTCAE: Common Terminology Criteria for Adverse Events; IRR: injection-related reaction; IV, intravenous; MS: multiple sclerosis.
In contrast to ocrelizumab, in vitro studies indicate that B-cell depletion by ofatumumab is mediated to a greater degree by complement-dependent cytotoxicity than by antibody-dependent cellular cytotoxicity.25,26 Furthermore, animal models have demonstrated that SC administration results in a prolonged release and absorption profile, primarily via the lymphatic system, enabling gradual interaction with B cells.27,28 The distinct pharmacokinetic and pharmacodynamic properties of ofatumumab may account for the efficacy of ofatumumab on B-cell depletion and clinical outcomes, utilizing a low dose and SC formulation, and could explain its positive tolerability profile.8,9,11,12
Consistent with other anti-CD20 B-cell-depleting therapies, the incidence of IRRs was highest following the first dose compared with subsequent doses, with no substantial difference between the continuous and newly switched ofatumumab groups.22–24 At the discretion of the trial clinician, patients were prescribed steroids, non-steroidal over-the-counter drugs or no pre-medication prior to ofatumumab injection, and medication was predominantly given prior to first injection. The tolerability analysis suggests a minimal impact of steroidal pre-medication on the reduction of systemic or local IRR incidence and severity, compared with the non-steroidal or no pre-medication groups. Systemic IRRs were predominantly mild and occurred after the first injection in all pre-medication categories. Steroid-associated AEs overlapped with ofatumumab systemic IRRs, which could have confounded AE reporting, yet the incidence of IRRs was not clinically different. If experienced, IRRs were predominantly mild and most commonly included a fever, which could be treated through utilizing non-steroidal antipyretics such as acetaminophen after ofatumumab administration, if required. Because pre-medication was given at the discretion of the trial clinician, the potential for bias cannot be excluded. 6
The lack of need for IV steroid pre-medication during ofatumumab treatment, and its good tolerability and safety profile may facilitate DMT access and independent management of RMS by patients.11,29 Interim analyses from patient-satisfaction research highlights that a DMT with convenient administration that fits “seamlessly into their lifestyle” and does not induce side effects that “interfere with daily life” is of great importance. Ofatumumab is an at-home, auto-injected DMT, with a small dose volume, enabling convenient self-administration, 8 which may reduce the need for IV-administration infrastructure and time burdens to receive care for patients. Furthermore, SC administration is preferred by patients, compared with IV delivery, 30 further supported by the high patient compliance with ofatumumab in this study. However, common reasons for injectable DMT non-adherence include injection anxiety and difficulty with self-administration; 21 thus, patients need sufficient support and education.
In conclusion, data from the overall safety population and post-marketing surveillance suggest that ofatumumab has a favourable tolerability profile. Formal pre-medication to reduce IRRs is not required and, if experienced, systemic IRRs are predominantly mild and experienced after the first injection. Ofatumumab allows at-home management of RMS to reduce the treatment burden for patients and HCPs.
Supplemental Material
sj-docx-1-mso-10.1177_20552173231203816 - Supplemental material for Tolerability of subcutaneous ofatumumab with long-term exposure in relapsing multiple sclerosis
Supplemental material, sj-docx-1-mso-10.1177_20552173231203816 for Tolerability of subcutaneous ofatumumab with long-term exposure in relapsing multiple sclerosis by John Kramer, Ralf Linker, David Paling, Adam Czaplinski, Olaf Hoffmann, V Wee Yong, Noreen Barker, Amy Perrin Ross, Elisabeth Lucassen, Mohammad Gufran, Xixi Hu, Ronald Zielman, Gustavo Seifer and Patrick Vermersch in Multiple Sclerosis Journal – Experimental, Translational and Clinical
Footnotes
Acknowledgements
The authors thank the patients for their participation in and commitment to the ASCLEPIOS I/II, APLIOS, APOLITOS and ALTHIOS trials and to the clinical study teams for the conduct of this study. The authors are grateful to Rachael Chandler, PhD, and Joseph Hawkins, PhD, of Oxford PharmaGenesis, Oxford, UK for providing medical writing support. These services were funded by Novartis Pharma AG.
Authors’ contributions
John Kramer: Trial design, data collection or responses to queries, data analysis, data review; manuscript review for intellectual content; final manuscript approval. Ralf Linker: Trial design, data collection or responses to queries, data analysis, data review; manuscript review for intellectual content; final manuscript approval. David Paling: Data collection or responses to queries, data review, manuscript review for intellectual content; final manuscript approval. Adam Czaplinski: Trial design, data collection or responses to queries, data analysis, data review; manuscript review for intellectual content; final manuscript approval. Olaf Hoffmann: Trial design, data collection or responses to queries, data analysis, data review; manuscript review for intellectual content; final manuscript approval. V. Wee Yong: Trial design, data collection or responses to queries, data analysis, data review; manuscript review for intellectual content; final manuscript approval. Noreen Barker: Trial design, data collection or responses to queries, data analysis, data review; manuscript review for intellectual content; final manuscript approval. Amy Perrin Ross: Trial design, data collection or responses to queries, data analysis, data review; manuscript review for intellectual content; final manuscript approval. Elisabeth Lucassen: Trial design, data collection or responses to queries, data analysis, data review; manuscript review for intellectual content; final manuscript approval. Mohammad Gufran: Trial design, data collection or responses to queries, data analysis, data review; manuscript review for intellectual content; final manuscript approval. Xixi Hu: Trial design, data collection or responses to queries, data analysis, data review; manuscript review for intellectual content; final manuscript approval. Ronald Zielman: Trial design, data collection or responses to queries, data analysis, data review; manuscript review for intellectual content; final manuscript approval. Gustavo Seifer: Trial design, data collection or responses to queries, data analysis, data review; manuscript review for intellectual content; final manuscript approval. Patrick Vermersch: Trial design, data collection or responses to queries, data analysis, data review; manuscript review for intellectual content; final manuscript approval
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship and/or publication of this article: JK has received consulting fees from Biogen, Genentech, Novartis, TG Therapeutics and Sanofi and has received research funding from Biogen and Genentech. OH's institution has received research funding from Biogen, Novartis and Sanofi. OH has received compensation for consulting and/or speaking activities from Alexion, Bayer, Biogen, Boehringer, Celgene, Janssen, Merck, Novartis, Roche, Sandoz and Sanofi. NB has received compensation for attending educational events and advisory boards, and for teaching from Biogen, Merck, Novartis, Roche and Sanofi. PV has received honoraria and contributions to meetings from Biogen, Sanofi-Genzyme, Novartis, Teva, Merck, Roche, Imcyse, AB Science, Almirall and Bristol-Myers Squibb-Celgene. PV has received research support from Novartis, Sanofi-Genzyme and Roche.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Novartis Pharma AG (Basel, Switzerland).
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.