
Abstract
Background:
Fabry disease is a rare disorder caused by the deficient activity of α-galactosidase A (GLA) that often leads to organ damage. Fabry disease can be treated with enzyme replacement or pharmacological therapy, but due to its rarity and nonspecific manifestations, it often goes undiagnosed. Mass screening for Fabry disease is impractical; however, a targeted screening program for high-risk individuals may uncover previously unknown cases.
Objective:
Our objective was to use population-level administrative health databases to identify patients at high risk of Fabry disease.
Design:
Retrospective cohort study.
Setting:
Population-level health administrative databases housed at the Manitoba Centre for Health Policy.
Patients:
All residents of Manitoba, Canada, between 1998 and 2018.
Measurements:
We ascertained the evidence of GLA testing in a cohort of patients at high risk of Fabry disease.
Methods:
Individuals without a hospitalization or prescription indicative of Fabry disease were included if they had evidence of 1 of 4 high-risk conditions for Fabry disease: (1) ischemic stroke <45 years of age, (2) idiopathic hypertrophic cardiomyopathy, (3) proteinuric chronic kidney disease or kidney failure of unknown cause, or (4) peripheral neuropathy. Patients were excluded if they had known contributing factors to these high-risk conditions. Those who remained and had no prior GLA testing were assigned a 0% to 4.2% probability of having Fabry disease depending on their high-risk condition and sex.
Results:
After applying exclusion criteria, 1386 individuals were identified as having at least 1 high-risk clinical condition for Fabry disease in Manitoba. There were 416 GLA tests conducted during the study period, and of those, 22 were conducted in individuals with at least 1 high-risk condition. This leaves a screening gap of 1364 individuals with a high-risk clinical condition for Fabry disease in Manitoba who have not been tested. At the end of the study period, 932 of those individuals were still alive and residing in Manitoba, and if screened today, we expect between 3 and 18 would test positive for Fabry disease.
Limitations:
The algorithms we used to identify our patients have not been validated elsewhere. Diagnoses of Fabry disease, idiopathic hypertrophic cardiomyopathy, and peripheral neuropathy were only available via hospitalizations and not physician claims. We were only able to capture GLA testing processed through public laboratories. Patients identified to be at high risk of Fabry disease by the algorithm did not undergo GLA testing due to a clinical rationale that we were unable to capture.
Conclusions:
Administrative health databases may be a useful tool to identify patients at higher risk of Fabry disease or other rare conditions. Further directions include designing a program to screen high-risk individuals for Fabry disease as identified by our administrative data algorithms.
Introduction
Fabry disease is an X-linked lysosomal storage disorder caused by the deficient activity of α-galactosidase A (GLA). This leads to the accumulation of its substrates, predominately globotriaosylceramide (GL-3) in lysosomes of tissues, resulting in cellular dysfunction that affects multiple organs. 1 Classic Fabry disease is a burdensome and life-limiting disease, shortening life-expectancy in affected individuals by an average of 17 to 20 years. 2 Affected patients are at high risk of developing peripheral neuropathy, progressive proteinuric chronic kidney disease (CKD), kidney failure, fibrotic cardiac disease, progressive hypertrophic cardiomyopathy, and ischemic stroke. 3
An early diagnosis of Fabry disease is essential so that enzyme replacement or pharmacological chaperone therapy can be initiated, with the aim of limiting potentially irreversible organ damage. Symptoms of Fabry disease are often experienced in early childhood, but diagnosis is frequently delayed until adolescence or early adulthood due to its rarity and the nonspecific nature of its manifestations. It is likely that many individuals with mild or late-onset disease are never tested as their symptoms are attributed to other diagnoses. 4 The condition is diagnosed in men by GLA activity testing in blood leucocytes. In affected women, random X-inactivation may result in the expression of GLA activity in the plasma or leucocytes within the normal range. Hence, genetic testing for the GLA gene is necessary for a diagnosis of Fabry disease in women.5,6 Furthermore, identification of a GLA variant is insufficient for a diagnosis of Fabry disease in heterozygous individuals in whom a pathogenic GLA variant is also required. 7
The prevalence of Fabry disease is estimated to be between 1 in 40 000 and 1 in 120 000 individuals. 8 These rates reflect clinically driven diagnoses; that is, testing is performed when symptoms are present or when the disease is suspected based on the familial pedigree. However, recent screening studies show that the prevalence of Fabry disease may be markedly higher than that previously estimated.9 -13 Acknowledging that many of these Fabry gene variants are of unclear significance, this discordance between the observed frequency of clinical disease and gene mutations admits the possibility that an uncertain proportion of Fabry disease patients may remain undiagnosed.
Several clinical scenarios have been associated with a higher frequency of Fabry disease including ischemic stroke in a young patient,14,15 idiopathic hypertrophic cardiomyopathy,16,17 proteinuric CKD or kidney failure of unknown cause,18,19 and peripheral neuropathy of unknown cause.20,21 These scenarios are associated with up to a 4% probability of Fabry disease. Therefore, screening for Fabry disease would be reasonable in these high-risk populations; however, it is not commonly performed. As a result, there may exist a large screening gap, representing missed opportunities for diagnosis and treatment. Our objective was to identify patients at high risk of Fabry disease in Manitoba, Canada, using population-level administration data and to estimate the proportion of patients at risk who were actually tested for Fabry disease.
Methods
Data Sources
We performed a retrospective cohort study using linked population-level administrative databases from Manitoba, Canada, provided by the Manitoba Centre for Health Policy (MCHP) at the University of Manitoba.22 -24 The MCHP databases have been extensively audited, validated, and used for prior studies. Information pertaining to unique individuals across various databases was linked through a de-identified version of the patient’s Personal Health Identification Number, a unique 9-digit number for each resident of Manitoba. Ethics approval for the study was provided by the University of Manitoba Health Research Ethics Board (file #: HS23595).
The administrative databases used included (1) Manitoba Health Insurance Registry: health insurance coverage dates and patient demographics; (2) Medical Services and Claims: billings for physician visits and services; (3) Canadian Institute for Health Information Discharge Abstract Database: inpatient hospital admissions, day surgeries, and discharge diagnoses; (4) Diagnostic Services of Manitoba (DSM): laboratory test results; and (5) Drug Program Information Network: outpatient drug prescriptions.
Study Design and Population
The study included all patients residing in Manitoba from January l, 1998, to December 31, 2018. We then stratified patients into 2 groups: (1) those who had evidence of existing Fabry disease and (2) those without any evidence of Fabry disease. An individual was identified as a Fabry disease case if they had a hospitalization during the study period with 1 of 2 specific International Classification of Diseases (ICD) diagnostic codes as either a primary or underlying diagnosis (ICD-9 code 272.2 or ICD-10 code E75.2) or if they had a prescription for enzyme replacement therapy (agalsidase alfa or agalsidase beta) or pharmacological chaperone therapy (migalastat). Prescriptions were identified via the following drug identification numbers: 02248965, 02248966, 02249057, 02468042.
In the group of patients without any hospitalization record for Fabry disease, we created a cohort who had at least one of the following high-risk comorbidities: ischemic stroke <45 years of age, idiopathic hypertrophic cardiomyopathy, kidney failure or proteinuria of unknown cause, and peripheral neuropathy (Appendix 1). We excluded patients with known contributing factors for these high-risk conditions by assessing laboratory tests, prescription drugs, and ICD diagnostic codes from physician billing and hospitalizations (Appendix 2). For example, for ischemic stroke <45 years of age, we first gathered a group of patients with a diagnosis of ischemic stroke and then excluded different groups of patients: age ≥45 years, diabetes, hypertension, autoimmune disease, and cancer, until we had a final list of patients. The known contributing factors used for exclusion criteria were assessed at any time before the first occurrence of a diagnosis of a high-risk condition, and up to 1 year after the first occurrence of a diagnosis of a high-risk condition. Those who remained and did not have evidence of prior GLA testing facilitated through DSM laboratories were assigned a probability of having Fabry disease based on estimates from the literature.
We used 2 systematic reviews to estimate the prevalence of Fabry disease in patients with stroke at a young age, idiopathic hypertrophic cardiomyopathy, and kidney failure.19,25 For stroke at a young age, the probability of Fabry disease was estimated to be from 0.1% to 4.2% in men and 0.1% to 2.1% in women. In individuals with idiopathic hypertrophic cardiomyopathy, the probability of Fabry disease was estimated to range from 0.7% to 2.1% in men and 0.7% to 2.3% in women. And in patients with kidney failure on hemodialysis, the probability of Fabry disease was estimated to range from 0.2% to 0.3% in men and 0% to 0.2% in women. For our probability estimates of peripheral neuropathy of unknown cause, we applied a 0% to 4.2% probability for both men and women based on lower and higher end estimates from 7 studies.20,21,26 -30
Laboratory Methods
For males, the method of GLA testing was to assess GLA activity measurement in serum by fluorometry. In females, GLA testing was done via gene sequencing to identify Fabry disease–causing mutations. In some cases, GLA activity was measured in leukocytes, and GL-3 was measured in urine tests.
Outcomes
Our primary outcomes were to determine the screening gap for Fabry disease and the potential number of missed Fabry disease cases in Manitoba, Canada. The screening gap was defined as the total number of patients who were considered to be at high risk of the disease subtracting those who have already been tested. In those patients with a screening gap, we assessed whether they had visited a cardiologist, nephrologist, or neurologist through specialist codes provided in the Medical Services and Claims data set. The potential number of missed Fabry disease cases was provided as a range and defined as the number of patients who comprised the screening gap multiplied by the lower probability estimate and higher probability estimate for each high-risk condition stratified by sex. For patients with Fabry disease, we reported the clinical incidence during the study period, and the distributions of age (mean and standard deviation) and sex (frequency and percentage). All analyses were performed using SAS 9.4 (SAS Institute, Inc., Cary, North Carolina).
Results
Patients With Fabry Disease
Throughout the study period, 2 063 280 instances of health insurance coverage were registered in the Manitoba Health Insurance Registry for 1 949 633 individuals: 980 512 (50.29%) men and 969 121 (49.71%) women. During the study period, 4 941 637 hospital admissions were reported in 1 280 066 patients. Among those hospitalizations, 186 were for Fabry disease associated with 17 patients: 10 (58.82%) men and 7 (41.16%) women. The average age of men when Fabry disease was diagnosed was 29 years (±24.50), and the average age of women at the time of diagnosis was 36 years (±24.45). The 17 cases of Fabry disease in 1 949 633 individuals correspond to a clinical incidence of 1 in 114 684 over the study period. There were 0 patients found to have a prescription for enzyme replacement or pharmacological chaperone therapy.
Patients at Risk of Fabry Disease
We found 60 447 individuals with a history of ischemic stroke, 11 684 individuals with potential idiopathic hypertrophic cardiomyopathy, 72 207 individuals with proteinuria or kidney failure, and 524 individuals with peripheral neuropathy (Figure 1). Applying our exclusion criteria reduced these numbers to 204, 655, 391, and 138, respectively. After combining all 4 high-risk disease groups and removing duplicates, the total number of individuals with at least 1 high-risk clinical condition for Fabry disease in Manitoba was 1386 (729 men and 657 women).

Identifying patients at high risk of Fabry disease.
A total of 511 GLA tests were conducted in 416 unique patients (178 women and 238 men). The median age of these patients was 46 years (interquartile range: 18-57). In our cohort at high risk of Fabry disease, 22 (1.6%) patients had at least 1 GLA test. This leaves a screening gap of 1364 individuals with a high-risk clinical condition for Fabry disease in Manitoba who have not yet been tested. By the end of the study period, 1096 (79.1%) of these individuals had at least 1 visit with a cardiologist, 408 (29.4%) of these individuals had at least 1 visit with a nephrologist, 634 (45.7%) of these individuals had at least 1 visit with a neurologist, and 99 (7.1%) had no record of a visit with any of those specialties.
Of those 1364 individuals, 932 were still alive and residing in Manitoba (504 men and 428 women) at the end of the study period according to the Manitoba Health Insurance Registry. If those patients were screened, we would estimate a range of 3 to 18 previously undiagnosed cases that would test positive for Fabry disease (Tables 1 and 2).
Estimates of Undiagnosed Fabry Disease Cases Among Males.

Individuals with more than 1 high-risk condition were counted more than once.
Estimates of Undiagnosed Fabry Disease Cases Among Females.
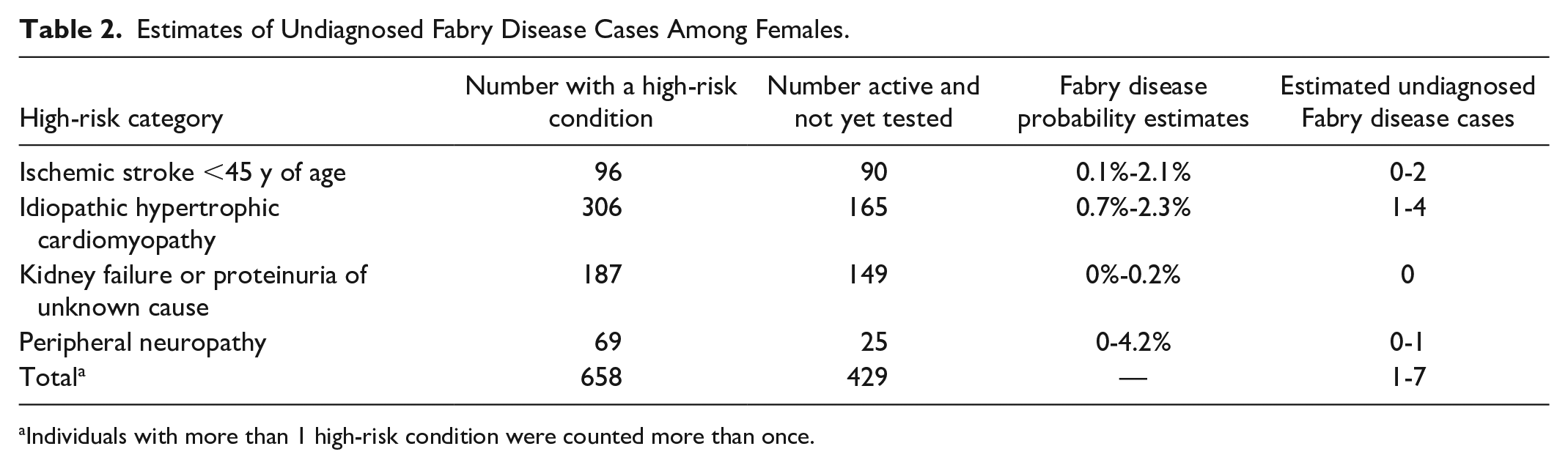
Individuals with more than 1 high-risk condition were counted more than once.
Discussion
In this retrospective cohort study of adults in Manitoba, we found an incidence of confirmed Fabry disease that is similar to clinical incidence rates that have been observed in other jurisdictions. 8 Throughout our study period, we found more than 1000 Manitobans at high risk who have not been screened for Fabry disease and had idiopathic hypertrophic cardiomyopathy, an ischemic stroke <45 years of age, kidney failure or proteinuria of unknown cause, or peripheral neuropathy without known contributing factors. This screening gap suggests a possibility, indicated by previous screening studies, that there may be a higher prevalence of Fabry disease in Manitoba than what is generally estimated based on the clinical diagnosis and pedigree analysis.9 -13 We found there may be between 3 and 18 individuals who have undiagnosed Fabry disease who could potentially benefit from enzyme replacement or pharmacological chaperone therapy if identified.
Because Fabry disease is so rare, a screening program for the general public would not be cost-effective as the number of new cases discovered would be too few relative to the costs of such a program. For a screening program to detect rare diseases to be cost-effective, it must be conducted in specific target population. Previous research has shown that screening for diseases in specific at-risk population could be cost-effective. 31 However, identifying patients who meet such criteria prospectively would be a challenge. Therefore, population-level administrative health databases, such as those held at the MCHP, can be a useful tool to identify these patients, and although the identity of the individuals cannot be known to researchers, there are tools available to have the Ministry of Health contact these patients for screening.
Large administrative data sets have been used in the past to identify rare diseases. Smith et al 32 developed an algorithm to find possible cases of muscular dystrophy through a linkage of private and public insurance programs and an all-payer hospital discharge data system in the United States and followed up to determine whether the algorithm correctly identified the possible cases. Of the 537 cases they found, 260 were determined to be true cases of muscular dystrophy compared to a nationwide frequency of 5.1 per 100 000. 33 In addition to ICD-9 and ICD-10 codes associated with muscular dystrophy, they also collected demographics, other neurological disorders, setting of care, provider specialties for all physician visits, and prescriptions of corticosteroids, which improved the probability that true cases of muscular dystrophy would be identified. This demonstrates the additional value administrative data can provide to screen for a rare disease and confirms the feasibility of using administrative data for that purpose.
Efforts to use large data sets to identify those at risk of Fabry disease are already underway. Jefferies et al 34 have developed an artificial intelligence (AI) tool following the predictive AI modeling methodology 35 to identify individuals with undiagnosed Fabry disease by using 4978 individuals with confirmed Fabry disease as a training data set to create their tool and tested their tool on a data set of 1 000 000 individuals without confirmed Fabry disease that was derived from various electronic medical records with health claims, medication history, and laboratory results. The researchers looked to see if the individuals the tool identified as high risk had a longitudinal medical history of various phenotypic signatures that medical experts would expect to see in patients with Fabry disease. They found that the tool showed very good discrimination with a c-statistic of 0.82 and projected a prevalence of Fabry disease of 1 in 2090 in the top 1% at-risk individuals without a confirmed disease, which is much higher than that in the general public. However, without actually screening these individuals, we cannot know for sure whether that prevalence is accurate, nor can we know which probability level assigned by the tool would be the appropriate cutoff to implement a cost-effective screening program. Our approach attempted to narrow the focus by identifying individuals with specific conditions known to be disproportionately associated with Fabry disease and excluded patients with known underlying causes of those conditions. This method may increase the probability that Fabry disease is the cause of those conditions, and therefore, we would expect a much high prevalence of true disease in our high-risk cohort than what was estimated in the top 1% at-risk individuals identified by the AI tool.
There have also been cohort studies in Canada with smaller samples sizes that have screened for Fabry disease in high-risk patients. A cohort of 397 patients with proteinuric CKD and at least 1 clinical characteristic for Fabry disease found 7 abnormal GL-3 test results in 4 men and 3 women, but none of those 7 individuals had confirmed Fabry disease after GLA testing. 36 However, in contrast to our analysis, they did not exclude patients with an explanation for proteinuric CKD. Therefore, we may expect to see more than 0 cases in our subgroup of high-risk patients with proteinuric CKD or kidney failure of unknown cause. In a single-center, hospital-based cohort of 100 patients aged 16 to 55 years who had a cryptogenic ischemic stroke, 1 patient was discovered to have Fabry disease due to a GLA mutation and high GL-3 levels, corresponding to a prevalence of 1% (95% CI: <.01%-6%). 37 Meanwhile, in a prospective multicenter Canadian cohort of 365 patients aged 18 to 55 years with a cryptogenic ischemic stroke or transient ischemic attack, 2% of patients screened positive for Fabry disease through GLA testing, but the prevalence was 0.3% when limiting to the p.R118C variant. 38 These ranges of confirmed Fabry disease are within what we have estimated we may find in our cohort of patients with a stroke <45 years of age.
This analysis has multiple strengths; as Canada has a universal health system, we were able to establish a true denominator for the incidence of Fabry disease in Manitoba and a comprehensive case definition to guide screening. In addition, we found a much higher percentage of previous GLA testing in our high-risk cohort than in those who did not meet our definition of being at high risk of Fabry disease (1.59% vs 0.02%), which is an indicator that the high-risk criteria chosen for this study are consistent with established clinical practice. However, there were limitations. The algorithms we used to identify our patients have not been validated elsewhere, and due to the specificity of the ICD-9 and ICD-10 codes, our algorithms were sometimes limited to data from reported hospitalizations only as we could not reliably search physician claims for Fabry disease, idiopathic hypertrophic cardiomyopathy, or peripheral neuropathy as these claims only document the first 3 digits of the ICD code for all diagnoses. Additionally, we were only able to capture proteinuria as well as GLA testing for genetic markers of Fabry disease processed through public laboratories. Private GLA testing arranged through manufacturers of enzyme replacement or pharmacological chaperone therapy would not have been captured, and as such, the true screening gap may be smaller than what is reported. Finally, there is a possibility that patients identified to be at high risk of Fabry disease by the algorithm did not undergo GLA testing due to a clinical rationale that we were unable to capture.
Future aims should test the feasibility and diagnostic yield of a comprehensive screening strategy for patients identified to be at high risk of Fabry disease in Manitoba. Our data show that 92.9% of patients identified to be at high risk of Fabry disease in our study have visited a specialist, and a future provincial screening program may be able to optimize case detection independent of care provider clinical impression, which may be a bias. For those who test positive, disease management should include genetic counseling, consideration of enzyme replacement or pharmacological chaperone therapy, organ-specific therapies (eg, renin-angiotensin-aldosterone system inhibition, cardiovascular risk reduction), and interdisciplinary care. Results from such an initiative will inform future strategies to improve the identification and outcomes of patients with Fabry disease worldwide. Future analyses could leverage these data to develop cost-effectiveness and willingness-to-pay threshold analyses for Fabry disease screen-and-treat strategies.
Conclusions
Fabry disease is a rare, multifaceted disorder that profoundly impacts the quality of life. Recent analysis and screening programs including this report have suggested that the condition may be underdiagnosed. Administrative health databases may be a useful tool to identify patients at higher risk of Fabry disease or other rare conditions. Further directions include designing a comprehensive screening program to identify previously undiagnosed cases of Fabry disease.
Supplemental Material
sj-docx-1-cjk-10.1177_20543581231162218 – Supplemental material for Magnitude of the Potential Screening Gap for Fabry Disease in Manitoba: A Population-Based Retrospective Cohort Study
Supplemental material, sj-docx-1-cjk-10.1177_20543581231162218 for Magnitude of the Potential Screening Gap for Fabry Disease in Manitoba: A Population-Based Retrospective Cohort Study by Reid H. Whitlock, Mohammad Nour-Mohammadi, Sarah Curtis, Paul Komenda, Clara Bohm, David Collister, Navdeep Tangri and Claudio Rigatto in Canadian Journal of Kidney Health and Disease
Footnotes
Acknowledgements
The authors acknowledge the Manitoba Centre for Health Policy for the use of data contained in the Manitoba Population Research Data Repository under project #2020-018 (HIPC#2019/2020-55). The results and conclusions are those of the authors, and no official endorsement by the Manitoba Centre for Health Policy, Manitoba Health, or other data providers is intended or should be inferred. Data used in this study are from the Manitoba Population Research Data Repository housed at the Manitoba Centre for Health Policy, University of Manitoba, and were derived from data provided by Manitoba Health and Shared Health Diagnostic Services.
Ethics Approval and Consent to Participate
Ethics approval for the study was provided by the University of Manitoba Health Research Ethics Board (file #: HS23595).
Consent for Publication
All authors have reviewed the manuscript and consented to publication.
Availability of Data and Materials
Data used in this article was derived from administrative health and social data as a secondary use. The data was provided under specific data sharing agreements only for approved use at Manitoba Centre for Health Policy (MCHP). The original source data is not owned by the researchers or MCHP and as such cannot be provided to a public repository. The original data source and approval for use has been noted in the acknowledgments of the article. Where necessary, source data specific to this article or project may be reviewed at MCHP with the consent of the original data providers, along with the required privacy and ethical review bodies. Requests to access to statistical and anonymous aggregate data associated with this paper, along with metadata describing the original source, can be made by contacting the corresponding author.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by Sanofi Genzyme Inc. The funding source had no role in the study design or interpretation of the results.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.