A Helix-Swapped C3d Dimer Mediated by Immune Evasion Protein Sbi Hints at a Novel S. Aureus Complement Modulation Strategy
Dunphy, R1, Wahid, A2, Back, C3, Martin, B4, Watts, A4, Dodson, C4, van den Elsen, J1
1Department of Biology and Biochemistry, University of Bath, Bath, United Kingdom
2Department of Pharmacology, University of Cambridge, Cambridge, United Kingdom
3School of Biochemistry, University of Bristol, United Kingdom
4Department of Pharmacy and Pharmacology, University of Bath, United Kingdom
Background: When Staphylococcus aureus enters the bloodstream, it acts as an opportunistic pathogen producing a range of immune evasion molecules which allow it to modulate complement activation. S. aureus binder of IgG (Sbi) can interact directly with central complement component C3 and its associated regulators. Recently we reported on the potential role of C3d dimers in modulating B-cell activation and here we show that Sbi can stabilise these C3d dimers by inducing an N-terminal helix swap, indicating a novel role in immune evasion.
Materials and methods: The C3d and Sbi proteins were recombinantly expressed and purified. The complex of dimeric C3d and Sbi IV was determined, using X-ray crystallography. The N-terminal helix swap observed in the crystal structure was further investigated by circular dichroism (CD) and fluorescence spectroscopy.
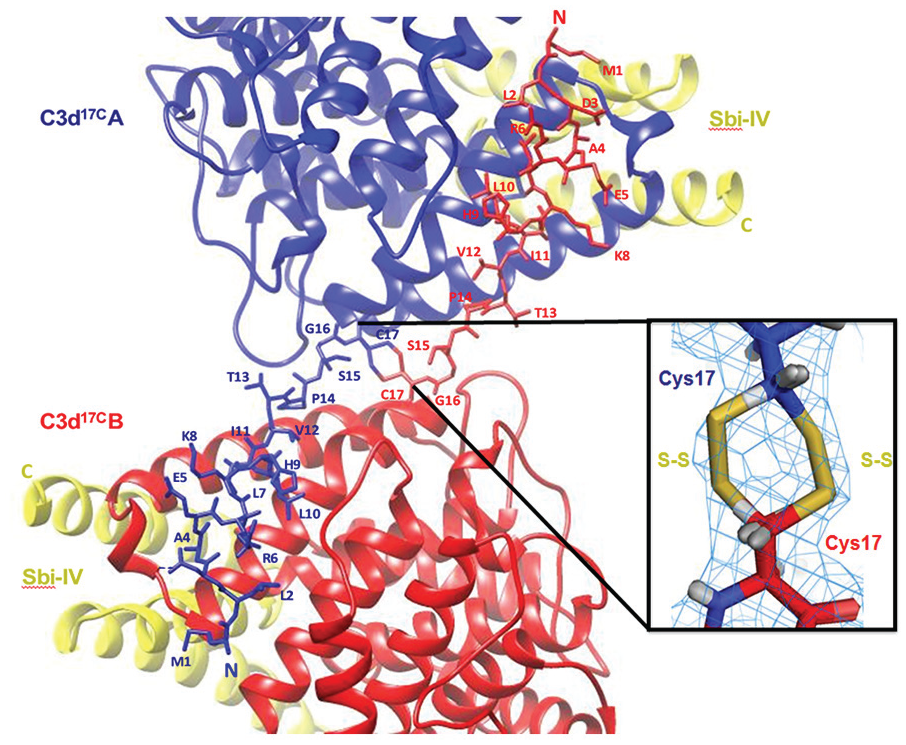
Results: The crystal structure of a C3d dimer Sbi-IV complex (Figure 1) reveals a unique N-terminal helix swap. The helix swap results in a significant increase in C3d dimer surface area which structurally stabilises the dimer, as was shown by CD spectroscopy analysis. The mechanistic aspects of the Sbi-induced helix swap were further elucidated using fluorescence spectroscopy.
Ribbon representations of an N-terminal helix-swapped human C3d17C dimer in complex with Sbi-IV at 2.4 Å resolution.
Discussion: Combined with our recent findings about the potential role of these dimers in B-cell modulation, our results suggest a new immune evasion strategy for S. aureus and could inform the design of novel therapies for immune system disorders in the future.
Conclusion: Structure-function analyses show that S aureus immune evasion protein Sbi can stabilise C3d dimers by inducing a unique N-terminal helix swap.
References
Serruto D, Rappuoli R, Scarselli M, Gros P, van Strijp JAG. “Molecular mechanisms of complement evasion: learning from staphylococci and meningococci”. Nature Reviews Microbiology. 2010 June; 8:393-399. doi: 10.1038/nrmicro2366
Wahid, A. A., Dunphy, R. W., Macpherson, A., Gibson, B. G., Kulik, L., Whale, K., Back, C., Hallam, T. M., Alkhawaja, B., Martin, R. L., Meschede, I., Laabei, M.,Lawson, A. D. G., Holers, V. M., Watts, A. G., Crennell, S. J., Harris, C. L., Marchbank, K. J., & van den Elsen, J. M. H. (2021). Insights Into the Structure- Function Relationships of Dimeric C3d Fragments. Frontiers in Immunology, 0, 3150. https://doi.org/10.3389/FIMMU.2021.714055
The Role of Factor H in Macrophages
Alexander, J1.
1Department of Medicine, University at Buffalo, Buffalo, NY 14203
Introduction: Complement factor H (FH) is a critical regulator of the alternative pathway. Dysfunctional or deficient FH underlie kidney diseases such as dense deposit disease and atypical hemolytic uremic syndrome. Macrophages are phenotypically heterogeneous, functionally diverse and highly plastic immune cells that play an important role in these diseases. Although FH is known to be a serum protein more recently it was shown to be present intracellularly in other cells. However, the role of FH at different locale is yet to be elucidated.
Methods: To investigate the role of FH in macrophages, bone marrow derived macrophage cultures were established from wildtype and FH deficient (FH-/-) mice. Different techniques such as Gene expression analysis, Sea horse etc for analyzing the cells.
Results: Wildtype(WT) macrophages in culture express FH and have a significantly higher M1/M2 ratio than FH-/- macrophages. FH deficiency reduced phagocytic activity in macrophages. Seahorse analysis showed reduced basal mitochondrial respiration, ATP production and maximal respiratory capacity in FH deficient macrophages, which is not affected by serum treatment or C3a and C5a, proteins generated by complement activation. Absence of FH showed a marked inability to form stable lamellipodia necessary for directional locomotion. Nanostring gene expression analysis revealed that the FH dependent gene changes in mouse macrophages include upregulation of inflammatory genes such as CX3CR, CXCR, IL-6 and TNFa, while genes such as antifibrotic MMP19 and antiproliferative BMP6 are reduced, which was substantiated by qRT-PCR.
Conclusion: For the first time, our studies show the functional relevance of intrinsic FH in macrophages.
An Antibody Targeting Complement Factor H Causes Anti-Tumor Immunity Through B-Cell Activation
1Department of Radiology, Duke University Medical Center, Durham, NC, USA
2Department of Immunology, Duke University Medical Center, Durham, NC, USA
3Department of Pharmacology and Cancer Biology, Duke University Medical Center, Durham, NC, USA
Background: We previously discovered antibodies to complement factor H (CFH) in early-stage, non- metastatic lung cancer patients (1). B cells from CFH antibody-positive individuals were isolated by affinity to an epitope-containing peptide, and a CFH-specific antibody (GT103) was identified as a candidate therapeutic. GT103 has tumor-specificity, causes complement activation and cytotoxicity of tumor cells, and has anti-tumor activity in mice (2, 3). We hypothesize that GT103 causes B cell activation through the interaction between activated complement fragments and the CR2 receptor (4, 5).
Materials and methods: Immune profiling and single cell RNA sequencing (scRNA-seq) of GT103-treated murine CMT167 tumors were performed. Flow cytometry was used to measure C3b/iC3b on human tumor cells and phospho-Syk on B cells following GT103 treatment. Immunofluorescence was used to visualize C3b/iC3b/C3c in GT103 treated CMT167 tumors. B cell depletion studies were performed using the CMT167 model.
Results: ScRNA-seq showed that GT103 causes significant increased gene expression in B cell activation pathways. C3b/iC3b levels increase on the surface of human tumor cells treated with GT103 (Fig 1), and levels of C3b/iC3b/C3c increase in CMT167 tumors treated with GT103. Phospho-Syk, a B cell activation marker, increases following GT103 treatment of tumor cells co- incubated with B cells in vitro. B cell depletion confirms GT103 causes anti-tumor immunity through B cells.
GT103 (200 µg/mL) + NHS (10%) treatment of H460 tumor cells causes increased C3b/iC3b. Heat inactivated NHS and non-specific IgG3 control antibody were used as negative controls. Cells were treated for 1 hr at 37°C. Each condition was done in triplicate. NHS + GT103 vs NHS + IgG3 p value = 0.02
Discussion: In addition to the importance of B cells, immune profiling of GT103-treated tumors showed changes in MDSCs, Tregs, and dendritic cells; these areas are currently being investigated.
Conclusion: GT103 mediated complement activation causes B cell activation in tumors.
References
Amornsiripanitch, N., Hong, S., Campa, M. J., Frank, M. M., Gottlin, E. B., & Patz, E. F., Jr. (2010). Complement factor H autoantibodies are associated with early stage NSCLC. Clin Cancer Res, 16(12), 3226-3231. doi:10.1158/1078-0432.CCR-10-0321
Campa, M. J., Gottlin, E. B., Bushey, R. T., & Patz, E. F., Jr. (2015). Complement Factor H Antibodies from Lung Cancer Patients Induce Complement-Dependent Lysis of Tumor Cells, Suggesting a Novel Immunotherapeutic Strategy. Cancer Immunol Res, 3(12), 1325-1332. doi:10.1158/2326-6066.CIR-15-0122
Bushey RT, Moody MA, Nicely NL, Haynes BF, Alam SM, Keir ST, Bentley RC, Roy Choudhury K, Gottlin EB, Campa MJ, Liao HX, Patz EF Jr. (2016). A Therapeutic Antibody for Cancer, Derived from Single Human B Cells. Cell Rep, 15(7), 1505-1513. doi:10.1016/j.celrep.2016.04.038
Toapanta, F. R., & Ross, T. M. (2006). Complement-mediated activation of the adaptive immune responses: role of C3d in linking the innate and adaptive immunity. Immunol Res, 36(1-3), 197-210. doi:10.1385/IR:36:1:197
Carroll, M. C., & Isenman, D. E. (2012). Regulation of humoral immunity by complement. Immunity, 37(2), 199-207. doi:10.1016/j.immuni.2012.08.002
C5aR2 Deficiency Ameliorates Inflammation in Antibody Transfer Experimental EPIDERMOLYSIS BULLOSA Acquisita and Suggests Regulating Action on the Decisive C5a Receptor 1
1Institute for Systemic Inflammation Research (ISEF, University of Lübeck, Lübeck, Germany)
2Center for Research on Inflammation of the Skin (CRIS, University of Lübeck, Lübeck, Germany)
3Department of Dermatology (University of Lübeck, Lübeck, Germany)
4Lübeck Institute of Experimental Dermatology (LIED, University of Lübeck, Lübeck, Germany)
5Division of Immunobiology (Cincinnati Children’s Hospital Medical Centre, University of Cincinnati College of Medicine, Cincinnati, Ohio, USA)
Background:Epidermolysis bullosa acquisita (EBA) is a rare blistering skin disease induced by autoantibodies directed against type VII collagen (COL7) [1,2]. Transfer of antibodies against murine COL7 (mCOL7) into mice mimics the effector phase of EBA and results in a subepidermal blistering phenotype [3,4]. Activation of the complement system and especially the C5a/C5aR1 axis and subsequent activation of neutrophils are critical for EBA pathogenesis [5,6]. However, the role of the second C5a receptor, C5aR2, in the pathogenesis of EBA is still elusive.
Methods: We sought to delineate the functional relevance of C5aR2 in the effector phase of EBA by injecting anti-mCOL7 antibodies into C5aR2-deficient (C5ar2-/-) and wild-type control mice. To decipher the molecular mechanisms mediated by C5aR2, neutrophils from C5ar2-/-, C5ar1-/- and wild-type control mice were activated in vitro and then their biological response was analyzed.
Results: In our mouse model, C5ar2-/- mice showed an attenuated disease phenotype, similar to C5ar1-/- mice, suggesting a pathogenic contribution of C5aR2. In addition, neutrophils from C5ar2-/- mice exhibited significantly lower C5a-mediated activation and migration in vitro. Both functions are completely absent when C5ar1-/- neutrophils are activated.
Discussion: Our in vitro-data suggest that C5aR2 might influences C5a-mediated neutrophil activation and migration by enhancing C5aR1 signaling. This is further supported by our in vivo-study, where C5aR2 deficiency ameliorated disease development. An effect that leads to complete disease protection when C5aR1 is missing[5,6].
Conclusion: Collectively, we demonstrate here a pro-inflammatory contribution of C5aR2 to the pathogenesis of (auto-)antibody-induced tissue damage in experimental EBA, which might be mediated by enhancing C5aR1 signaling.
References
Koga et al., Front Med 2019;5:362.
Schmidt and Zillikens, Lancet 2013;381(9863):320-32.
Sadik et al., J Allergy Clin Immunol 2020;145(4):1145-47.
Sadik et al., Semin Immunol 2018;37:21-29.
Karsten et al.; Nat Med 2012;18(9):1401-6.
Mihai et al., Front Immunol 2018;9:535.
Clinical and Biomarker Characteristics of Patients with C3G Enrolled in Two Phase 2 Studies Investigating the Factor D Inhibitor Danicopan
2Molecular Otolaryngology and Renal Research Laboratory, Iowa, USA
3Columbia University Medical Center, New York, USA
4ZNA Nierkliniek, Antwerp, Belgium
5Imperial College London, London, UK
6Istituto di Ricerche Farmacologiche Mario Negri IRCCS, Bergamo, Italy
7Renal Section, Department of Pediatrics, University of Colorado School of Medicine, Colorado, USA
8Feinberg School of Medicine, Northwestern University, Illinois, USA
9Centre for Inflammatory Disease, Dept of Immunology and Infection, Faculty of Medicine, Imperial College London, London, UK
10Alexion Pharmaceuticals, Boston, MA, USA
11Division of Nephrology, The Ohio State University Medical Center, Ohio, USA
12Division of Immunology and Inflammation, Department of Medicine, Imperial College London, UK
13Johns Hopkins University School of Medicine, Maryland, USA
14NYU Grossman School of Medicine, New York, USA
15Emory University School of Medicine, Georgia, USA
16Department of Nephrology, Radboud University Medical Center Geert, Netherlands
Background: C3 glomerulopathy (C3G) is a rare progressive kidney disease attributed to dysregulation of the complement alternative pathway (AP). We describe baseline biomarker and clinical characteristics of C3G patients in two phase II studies of the oral factor D inhibitor danicopan (ALXN2040).
Materials and methods: NCT03369236 was a double-blind study of patients with C3G treated with danicopan or placebo. NCT03459443 was a single-arm, open-label study of patients with C3G or IC- MPGN treated with danicopan. All patients were to have proteinuria ≥500 mg/day and eGFR ≥30 mL/min/1.73 m2. Selected complement biomarkers were measured in serum, plasma, or urine prior to dosing.
Results: 29 patients with biopsy-confirmed C3G were included in this analysis. Eighteen were male ([62.1%]) and median (IQR) age at baseline was 24.0 (10.0) years. Baseline clinical and biomarker data are shown in Table 1. Abnormalities in biomarkers of AP and terminal pathway were observed. Reduced systemic C3 was observed in most patients and correlated strongly with increased sC5b-9 and reduced C5 (Spearman correlations [rs]=-0.73 and 0.80 [p<0.0001]). Ba and FD elevations correlated with lower eGFR (rs=-0.83 and -0.87 [p<0.0001]). Urine sC5b-9 (normalized to creatinine) correlated with plasma sC5b-9 and with proteinuria (protein to creatinine) (rs=0.69 and 0.83 [p<0.0001]).
Baseline clinical characteristics and biomarkers data.
Biopsy-Confirmed C3G N=29
Total N=35
Disease duration (years)
Median (Q1, Q3)
5.45 (1.90, 7.65)
5.10 (2.00, 7.30)
eGFR (mL/min/1.73 m2)
Median (Q1, Q3)
81.200 (51.530, 117.800)
81.200 (51.530, 119.310)
Ratio of 24-hour urine protein/creatinine (mg/mmol)
Conclusions: Fluid phase biomarker assessments and correlations confirmed systemic terminal pathway activation associated with AP dysregulation in C3G patients. Ba and FD were strongly correlated with eGFR, confounding their use as markers of complement activation. Additional characterizations including autoantibodies and genetic variants will contribute further to our characterization of C3G pathophysiology.
Perceiver-Based Machine Learning Diagnosis of TMA in Renal Biopsies
1Institute of Pathology, University Hospital of Cologne, Cologne, Germany
2Department of Pathology, Weill Cornell Medicine, Cornell University, NY, USA
3Department of Pathology, Amsterdam UMC, Amsterdam, The Netherlands
4Center of Research of Immunopathology and Rare Diseases, University of Turin, Turin, Italy
5Biomedical Engineering & Medicine, University of Houston, Houston, TX, USA
6Electrical and Computer Engineering, University of Houston, TX, USA
7Department of Pathology, Leiden University Medical Center, Leiden, The Netherlands
Background: TMAs can show a wide spectrum of findings in renal biopsies. The role of nephropathology in the context of genetics and laboratory findings in the diagnosis, determination of etiology, prediction of therapy response and prognosis is unclear. Although clinicopathological registries could address these questions, large-scale nephropathology annotation seems too costly and prone to variation. In this proof-of-concept study, we propose a novel deep learning network architectures allowing multimodal data integration from the three decisive tissue compartments and the four routine nephropathological stainings.
Material and methods: 25 renal biopsies with a histopathological diagnosis of TMA and n=25 mimickers of TMA (ANCA-associated necrotizing arteritis, severe benign nephrosclerosis, cryoglobulinemic vasculitis, Bevacizumab-plugs) from two institutions were digitized. A total of 1366 arteries, 9561 arterioles and 7112 glomeruli from HE, PAS, trichrome and silver stainings at a 40x scan magnification were loaded into a novel network architecture including transformer and perceiver operations to train a binary classifier of TMA vs. Mimicker with five-fold internal cross-validation.
Results: So far, without hyperparameter tuning, we achieved a binary diagnostic accuracy of 82% through five-fold internal cross-validation.
Discussion: Our novel architecture can be trained to integrate histopathological findings from arteries, arterioles and glomeruli to render a diagnosis of TMA across the four common nephropathological stainings.
Conclusion: This novel architecture could be expanded with further input channels like tubulointerstitial tiles, EM images, genetic sequences and laboratory data to address open questions regarding TMAs. Together with powerful segmentation models this architecture enables the composition and examination of large clinicopathological registries.
Reccurence of Atypical Hemolytic Uremic Syndrome After COVID-19 Vaccination
1Laboratorio de Hemostasia y Trombosis, IMEX-CONICET-ANM, Ciudad Autónoma de Buenos Aires, Argentina.
2Clínica Bazterrica, Ciudad Autónoma de Buenos Aires, Argentina
3Departamento de Hemostasia y Trombosis, Instituto de Investigaciones Hematologicas Dr. Mariano R. Castex, Academia Nacional de Medicina, Ciudad Autónoma de Buenos Aires, Argentina.
Background: Atypical Hemolytic Uremic Syndrome (aHUS) is a complement-mediated thrombotic microangiopathy. Pathophysiological mechanism involves uncontrolled complement activation due to a genetic or acquired anomaly coupled with a triggering event. We report a case of aHUS recurrence following COVID-19 vaccination.
Material and methods: Whole blood (EDTA) was collected and processed with CD46-PE, CD45-PerCP, isotype control-PE markers. Staining was measured through median fluorescence intensity and expressed as CD46/isotype ratio. Sanger sequencing was used for identification of variants in CD46 gene. All the participants provided informed written consent.
Results: Proband (P) is a 39-year-old woman admitted for nausea, vomiting, epigastric pain and haematuria, three days after first dose of ChAdOx1 nCov-19 vaccine. Laboratory testing showed MAHA (Hb:8.8g/dL, Ht:26%), thrombocytopenia (80x109/mm3) and acute kidney injury (Cr:2.15mg/dL, Ur:92mg/dL). P and three of her siblings have experienced recurrent TMA episodes since childhood. In 2019, genetic study from P’s sister (S) identified two heterozygous variants in CD46, one pathogenic (Glu179Gln) and one of uncertain significance (Cys94Tyr). We demonstrated that P carries the same variants and observed a 50% decrease of CD46 expression in both P and S (fig.1).
Detection of CD46 on the surface of lymphocytes and neutrophils of proband (P), proband’s sister (S) and the control individual (healthy donor). A. Flow cytometric analysis using phycoerythrin (PE)-conjugated anti-CD46 was performed 24 hours after blood collection for the three samples. B. Staining was measured through median fluorescence intensity (MFI) and reference intervals (RI) were previously calculated using ratio CD46 MFI/isotype MFI in 20 control individuals. CD46 expression from P and S is decreased by approximatively 50% compared with median of reference samples.
Platelet transfusion, corticosteroids and 9 sessions of plasmapheresis contributed to rapid recovery of P.
Discussion: Glu179Gln was reported to increase CD46 expression on granulocytes in aHUS patient and to reduce C4b cofactor activity1. We observed that combination of Glu179Gln and Cys94Tyr was associated with low levels of CD46 on cell surface.
Conclusion: This case report supports the evidence of COVID-19 vaccine as a precipitating event for aHUS recurrence.
References
Frémeaux-Bacchi et al., J Am Soc Nephrol, 2006; 17: 2017–25.
Macrophage Depletion Reduces Disease Pathology in Factor H Dependent Immune Complex Glomerulonephritis
Shane Fraher1, Michael Phelps, Marcos Lopez, Alexander Jacob and Jessy Alexander
1Department of Medicine, University at Buffalo, Buffalo, NY 14203
Background/aims: Factor H (FH) is a critical regulator of the alternative complement pathway in both mice and men. Therefore, it is not surprising that FH abnormalities are associated with renal diseases. Macrophages (Mφs) play a critical role in FH dependent immune complex glomerulonephritis (ICGN), but the mechanism remains unclear.
Methods: In this study, ICGN was induced by immunizing 8-wk-old FH knockout (Cfh−/−) mice daily for 5 wk with 4 mg horse spleen apoferritin given ip. To determine the role of Mφs in this setting, during the last two weeks of apoferritin treatment, one group of mice received clodronate that selectively depletes Mφs in mice, while the other group received saline. Kidney pathology and function was assessed using different techniques.
Results: Gene expression analysis of kidneys Cfh−/− mice with ICGN revealed that a number of Mφ related genes were significantly elevated, compared to the kidneys from control mice. In mice, monocytes/Mφs were significantly reduced in circulation by seven days after clodronate administration. Chlodronate treated, ICGN mice had significantly lower levels of both serum creatinine (p<0.05) and BUN (p<0.02), compared to PBS/ICGN mice. In line with BUN and creatinine, histopathological kidney injury is significantly reduced. Chlodronate treatment did not significantly change C3 and IgG deposits, but reduced laminin in the kidney. The expression of Mφ inflammatory mediators such as ICAM, MMP9 and iNOS were reduced with chlodronate treatment.
Conclusion: Our results indicate an important role for macrophages in the pathogenesis of FH dependent ICGN, and implicate macrophages as a potential therapeutic target.
A Young Female with Cortical Necrosis Treated with Intravenous Immunoglobulin in a Resource Poor Setting: A Case Report
Monika K1, Nair S2, Balasubramanian K3
1DM Resident, department of Nephrology, Saveetha medical college, Chennai, India
2Professor and Head of department of Nephrology, Saveetha medical college, Chennai, India
3Associate Professor department of Nephrology, Saveetha medical college, Chennai, India
Background: Renal cortical necrosis is a rare cause of acute kidney injury (AKI) characterized by diffuse or patchy ischemic coagulation necrosis of cortex. One of important cause is Acute Thrombotic Microangiopathy (TMA). It is very crucial to diagnose & treat it early to avoid morbidity & mortality. Here we report a case of TMA with cortical infarct with poor response to plasmapheresis & steroid, treated with Intravenous Immunoglobulin (IVIG).
Material and methods: A 30-year female, presented with RPGN with anuria with with no extrarenal features of organ involvement. She found to have severe progressive anemia with mild thrombocytopenia with raised serum LDH, mild indirect hyperbilirubinemia with normal coagulation parameters with low C3, normal C4, immunological marker & APLA negative. Renal Biopsy done suggestive of acute TMA with cortical necrosis.
Results: Her urine output improved with 5 sessions of Plasmapheresis (PLEX), pulse steroid, followed by oral steroid. But her Renal parameters & anemia continued to worsening and she remained dialysis dependent. She received IVIG, to which she responded and currently she is off dialysis & hematological & renal parameters improving. Her complement gene analysis report is awaited.
Discussion: Anuria with dialysis dependency are bad prognostic markers for case of TMA with cortical necrosis, if there is delay in diagnosis or treatment. In a resource poor setting, we should avoid delay in treatment. In this case refractory to treatment to standard therapy got good response to IVIG.
Conclusion: In a case of Acute TMA with cortical infarct IVIG may help if there is partial or failure to response to standard therapy.
References
Prakash, J., & Singh, V. P. (2015). Changing picture of renal cortical necrosis in acute kidney injury in developing country. World journal of nephrology, 4(5), 480–486. https://doi.org/10.5527/wjn.v4.i5.480
Palma, L., Sridharan, M., & Sethi, S. (2021). Complement in Secondary Thrombotic Microangiopathy. Kidney international reports, 6(1), 11–23. https://doi.org/10.1016/j.ekir.2020.10.009
McFarlane, P. A., Bitzan, M., Broome, C., Baran, D., Garland, J., Girard, L. P., Grewal, K., Lapeyraque, A. L., Patriquin, C. J., Pavenski, K., & Licht, C. (2021). Making the Correct Diagnosis in Thrombotic Microangiopathy: A Narrative Review. Canadian journal of kidney health and disease, 8, 20543581211008707. https://doi.org/10.1177/20543581211008707
Blasco, M., Guillén, E., Quintana, L. F., Garcia-Herrera, A., Piñeiro, G., Poch, E., Carreras, E., Campistol, J. M., Diaz-Ricart, M., & Palomo, M. (2020). Thrombotic microangiopathies assessment: mind the complement. Clinical kidney journal, 14(4), 1055–1066. https://doi.org/10.1093/ckj/sfaa195
Ito, S., Okuyama, K., Nakamura, T. et al. Intravenous gamma globulin for thrombotic microangiopathy of unknown etiology. Pediatr Nephrol22, 301–305 (2007). https://doi.org/10.1007/s00467-006-0326-9
Clinical Characteristics of a Patient Population with Atypical Haemolytic Uraemic Syndrome and Malignant Hypertension: The Global aHUS Registry Analysis
1Division of Nephrology, The Hospital for Sick Children, Toronto, Ontario, Canada
2Service de Néphrologie-Hypertension Artérielle, Dialyses, Transplantation Rénale, CHRU Tours, France
3Alexion Pharmaceuticals Inc., Boston, United States
4Center for HUS Control, Prevention and Management, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Milano, Italia
5Department of Renal Medicine, Sir Charles Gairdner Hospital, Perth, Australia
6Department of Nephrology and Hypertension, Antwerp University Hospital, Edegem, Belgium
7Division of Pediatric Nephrology, Heidelberg University Hospital, Heidelberg, Germany
8Department of Internal Medicine and Pediatrics, Ghent University Hospital, Ghent, Belgium
9Urgences Néphrologiques et Transplantation Rénale, Hôpital Tenon, Paris
Background: Atypical haemolytic uraemic syndrome (aHUS) is a rare disease that manifests as complement-mediated thrombotic microangiopathy (TMA), which can lead to severe organ damage. Some patients with aHUS may present with malignant hypertension (MHT); both conditions can result in TMA. The objective of this analysis was to characterise patients with aHUS and MHT.
Materials and methods: In this analysis, patients from the Global aHUS Registry (NCT01522183) were included if they were diagnosed with MHT and were followed ≥90 days after initial aHUS symptom presentation or diagnosis date. Demographics and clinical characteristics were evaluated (table).
Clinical characteristics of patients with aHUS and MHT.
Characteristics
Statistics N=71
Age at aHUS diagnosis (years)
Median (min, max)
28.1 (0.5, 68.2)
Sex, M/F (n (%))
28 (39.4)/43 (60.6)
Eculizumab treatment duration (months), n
51
Median (min, max)
33.9 (0.1, 110.9)
Patients with kidney transplant, n
33
Patients treated with eculizumab in the peri- or post-transplant period, n (%)
20 (60.6)
Patients treated with eculizumab before transplant (> 1 year), n (%)
New extra-renal manifestations not present at time of initial aHUS symptoms
Cardiovascular, n (%)
19 (26.8)
Gastrointestinal, n (%)
15 (21.1)
Central nervous system, n (%)
13 (18.3)
Pulmonary, n (%)
9 (12.7)
Genetic mutation status
Any mutation found or anti-CFH-antibody positive, n (%)
40 (56.3)
Baseline biomarkers
Serum creatinine, µmol/L, (n, median, (min, max))
44 227 (21, 1330)
Platelet counts, x 103/µL, (n, median, (min, max))
40 202 (22, 455)
Lactate dehydrogenase, U/L, (n, median (min, max))
48 287 (134, 3332)
Before aHUS: more than 2 months prior to aHUS initial symptoms; After aHUS: more than 2 months after aHUS initial symptoms; Around the same time: aHUS initial symptoms ± 2 months.
Results: Seventy-one of 1903 registry patients were included in the analysis. Sixty-nine percent of patients were reported to have MHT at around the same time as aHUS diagnosis (+/-2 months), while 11% and 13% experienced MHT before and after aHUS diagnosis, respectively. aHUS triggering conditions were reported in 6/71 patients (8%) (Table).
Cardiovascular (27%) and gastrointestinal (21%) symptoms were the most commonly reported extra-renal manifestations. Eight patients (11%) had a reported family history of aHUS and 40 patients (56%) had a complement pathogenic variant or an anti-CFH-antibody. Thirty-three patients (46%) had a kidney transplant; of these, 20 were prescribed eculizumab in the peri- or post-transplant period.
Conclusion: In this analysis of patients with aHUS and MHT, the observed high prevalence of pathogenic variants in complement genes or anti-CFH antibodies, alongside the high proportion of patients with extrarenal manifestations and/or requiring kidney transplant, indicate a high severity of presentation and poor prognosis of aHUS associated with MHT.
Longitudinal sC5b-9 Levels and Atypical HUS Relapse in Two Pediatric Patients
1Pediatric Nephrology Unit, 1st Department of Pediatrics, Aristotle University of Thessaloniki, Hippokratio General Hospital, Thessaloniki, Greece
2Pediatric Nephrology Unit, 1st Department of Pediatrics, Aristotle University of Thessaloniki, Hippokratio General Hospital, Thessaloniki, Greece
3Hematology Department-BMT Unit, G. Papanicolaou Hospital, Thessaloniki, Greece
Background: sC5b-9 reflects complement cytolytic function. Its role as a biomarker of complement activation in atypical HUS has not been elucidated.
Material and methods: We report 2 cases of 6 and 7 year-old girls with atypical HUS attributed to single nucleotide polymorphisms in C3 (rs2230199) and CFH (rs800292) gene in the first case and in C3 (rs2230199) gene in the second case. Eculizumab therapy was initiated in both patients. sC5b-9 levels were measured, and HUS relapse episodes were recorded.
Results: sC5b-9 level was high at initial HUS presentation in both patients (333 ng/ml and 342 ng/ml respectively, normal range <245 ng/ml) and fell after eculizumab initiation (201 ng/ml and 275 ng/ml respectively). In the first patient, therapy was postponed at 3 and at 13 months after disease onset, leading to high sC5b-9 levels (913 ng/ml and 1020 ng/ml respectively), but clinical HUS relapse was recorded only during the first therapy postponement. In all cases, sC5b-9 levels fell to normal range after therapy re-initiation. In the second patient, therapy was postponed at 5 months after disease onset, leading to clinical HUS relapse, without rise in sC5b-9 levels (231 ng/ml).
Discussion: sC5b-9 levels rise and reduction after eculizumab postponement and re-initiation suggest that longitudinal sC5b-9 changes reflect complement activation. Nevertheless, we observed a variability of sC5b-9 levels during HUS relapse at both inter-individual and intra-individual level.
Conclusions: sC5b-9 may be useful for monitoring complement activation in atypical HUS in some patients but cannot predict HUS relapse at both inter-individual and intra-individual level
Atypical Hemolytic Uremic Syndrome Triggered by STEC Infection: A Case Report
1Pediatric Nephrology Unit, 1st Department of Pediatrics, Aristotle University of Thessaloniki, Hippokratio General Hospital, Thessaloniki, Greece
2Pediatric Nephrology Unit, 1st Department of Pediatrics, Aristotle University of Thessaloniki, Hippokratio General Hospital, Thessaloniki, Greece
3Hematology Department – BMT Unit, G. Papanicolaou Hospital, Thessaloniki, Greece
Background: The concomitant incidence of shiga-toxin Escherichia Coli (STEC) infection and atypical HUS has been rarely reported.
Material and methods: A 6-year-old girl was presented with HUS without vomiting or diarrhea. At 5th day of hospitalization, peritoneal dialysis therapy was started and lasted 20 days. During hospitalization, the patient presented pulmonary oedema and transient right ventricular diastolic dysfunction, necessitating an ICU stay for 2 days.
Results: At admission, fresh frozen plasma infusion therapy was initiated due to atypical clinical presentation and continued for 20 days. Of note, shiga-toxin 1 was detected at stool-PCR test, despite the absence of gut symptoms. ADAMTS13 activity was normal. Initial serum C5b-9 level was high (333 ng/ml, normal range<245 ng/ml). Further complement analysis revealed single nucleotide polymorphisms at both C3 (rs2230199) and complement factor H (rs800292) genes. Due to the observed complement activation dysregulation, eculizumab therapy was initiated and serum C5b-9 levels fell to normal range (201 ng/ml). The patient presented HUS relapse 3 months later, concomitantly to rise in C5b-9 levels (913 ng/ml), due to transient therapy postponement, which was rapidly resolved after eculizumab infusion. Serum C5b-9 levels gradually fell to normal range (159 ng/ml).
Discussion: In our case, asymptomatic STEC infection was associated with atypical HUS, suggesting that shiga-toxin may trigger uncontrolled complement activation in pediatric patients with inherited complement gene deficits.
Conclusion: Complement activity analysis needs to be included in the initial diagnostic work-up of HUS, even at the presence of documented STEC infection.
Utilizing Pharmacokinetic Studies to Optimize Therapy in a Child with Nephrosis and C3 Glomerulonephritis: A Precision Medicine Approach
1Division of Nephrology, The Hospital for Sick Children, Toronto, ON, Canada.
2Division of Pathology, The Hospital for Sick Children, Toronto, ON, Canada.
3Division of Pharmacology, The Hospital for Sick Children, Toronto, ON, Canada.
4Department of Paediatrics, University of Toronto, ON, Canada
Background: C3 glomerulonephritis (C3GN) is caused by complement alternative pathway dysregulation and is characterized by progression to ESRD. Terminal complement blockade via eculizumab has successfully been used especially in patients with elevated C5b-9 levels.
Results: We describe a 6 year old boy with C3GN, who presented with nephrotic syndrome, severe hypertension (on 4 anti-hypertensive medications) and acute kidney injury. Complement C3 level was 0.12 g/L (0.8-1.5) with pos C3NeF and elevated C5b-9 levels (2135, <239ng/ml). Despite 6 months of treatment with prednisone and MMF, he had ongoing nephrotic syndrome and worsening kidney function. He commenced on standard Eculizumab dosing for aHUS. Despite 6 months of therapy, he had persistent severe hypertension, nephrotic syndrome requiring weekly albumin/furosemide infusions and worsening kidney function. Complement C3 levels remained low with elevated C5b-9 levels, suggesting sub-optimal terminal complement inhibition due to urinary loss. We confirmed sub-therapeutic plasma concentrations of eculizumab as free plasma eculizumab levels were low on day 7 (9, >99 ug/ml) and undetectable on days 10 and 14 post-infusion. On the other hand, Eculizumab was detected in the urine, indicating urinary loss. He was subsequently increased to weekly eculizumab infusions and MMF dosing was adjusted using MPA AUC measurements. Since then, his kidney function, C5b-9 levels (297 ng/ml) and nephrotic syndrome improved significantly (see Figure), leading to discontinuation of albumin/furosemide and anti-hypertensive medications. (see Figure).
Overview of kidney function, proteinuria, C3 levels and MPA AUC since patient presented (Feb 2020). Vertical lines indicate start of Eculizumab Q14d and Eculizumab Q7d. Patient received a prednisone taper within first months and was started on MMF, which was increased to target an AUC >30mg*h/L.
Discussion: Use of pharmacokinetic studies can aid in individualized eculizumab treatment in those with C3GN who failed to respond to standard dosing, especially in patients with significant proteinuria as Eculizumab can be lost in the urine.
Dense Deposit Disease: An Overlap Syndrome
Maravi N1, Hassan A2, Bhat R3, Chennu K4
1Mazumdar shaw medical centre ( Departmet of Nephrology, Narayana health city, Bangalore, India)
2Mazumdar shaw medical centre (Department of Paediatric Nephrology, Narayana health city, Bangalore, India)
3Mazumdar shaw medical centre (Department of Nephrology, Narayana health city, Bangalore, India)
4Mazumdar shaw medical centre (Department of Nephrology, Narayana health city, Bangalore, India)
Introduction: Dense deposit disease (DDD) is a rare renal disease mostly characterized by an membranoproliferative pattern of injury with C3 accumulation and absent immunoglobulins. There can be an overlap between DDD, C3glomerulonephritis and atypical infection related glomerulonephritis.
Case description: We report 3 cases ofDDD, a 5 year and 19 yr old with similar symptoms of anasarca and hematuria; and a 23 yr old boy was treated as C3GN and later presented with worsening renal failure.
The child presented with persistent nephrotic features and hypocomplementaemia. Renal biopsy showed diffuse exudative glomerulonephritis and DDD on electronmicroscopy. He was treated with steroid and mycophenolate mofetil. His immunosuppression was upgraded to tacrolimus following a relapse. Currently he is doing well.
Second case of a teenager, had nephritic- nephritic features with renal biopsy showing C3GN and DDD on EM. He received standard course of steroid and on low dose steroid for maintainance. He is symptomatically well however his renal function is gradually worsening.
An young adult, symptomatic for four years and treated elsewhere with steroid, MMF and rituximab for a diagnosis of C3 glomerulonephritis. On his first visit to us, his clinical profile was suspicion of DDD. EM was suggestive of DDD and >50% interstitial fibrosis and tubular atrophy. Given the chroniciy, he is on conservative management.
Conclusion: All three cases highlights overlapping features of c3 glomerulonephritis. This emphasises the need to perform EM study to diagnose DDD, aggressively investigate to aid treatment plan and prognosticate.
Spectrum of Complement Factor H Related Gene Aberrations Observed in A Panel of Renal Diseases at a Tertiary Care Centre
Ray A1, Pal A2, Raychaudhury A3
1PDT,Department of Nephrology, IPGME&R & SSKM Hospital, Kolkata,INDIA
2Asst Prof, Department of Nephrology, IPGME&R & SSKM Hospital, Kolkata,INDIA
3Prof, Department of Nephrology, IPGME&R & SSKM Hospital, Kolkata,INDIA
Background: Basic research into complement biology has helped understand the pathogenesis of kidney disease hence,useful to consider the diseases under two broad categories; aHUS and C3GN. 60% cases of aHUS1 and 20% of C3GN2 are associated with mutations in components of alternative pathway, or anti CFH antibodies.
Materials & methods: Cases presenting with evidences of thrombotic microangiopathy (TMA) during the study period (Jan’20-Feb’21),were evaluated for pathologic variations in complement regulation via NGS based assays.In selected cases anti-CFH antibody titres were done.
Results: Among 10 patients,4 presented with suspected aHUS,2 – pregnancy related acute kidney injury (PRAKI) with TMA, rest 4 – acute glomerulonephritis (AGN),IC-MPGN, chronic TMA & CONS respectively.Cases fell within 6-34 yrs,median of 16.5 yrs & F:M ratio of 1.5:1.
Discussion: The probands with high anti CFH titres signified severe disease,we resorted to PLEX and cyclophosphamide3 therapy in both but one expired eventually.MMF started as maintainence therapy in the other.
Conclusion: Therefore, early diagnosis of pathogenic variants and initiation of PLEX along with immunosuppressive therapy can result in delaying ESRD in complement mediated diseases & also can help in shaping up future transplants in appropriate time.
References
Kavanagh D, Goodship T. Genetics and complement in atypical HUS. Pediatr Nephrol 2010; 25: 2431–2442.
Servais A, Noel LH, Roumenina LT et al. Acquired and genetic complement abnormalities play a critical role in dense deposit disease and other C3 glomerulopathies. Kidney Int 2012; 82:454–464
1Department of Immunology, Duke University Medical Center, Durham, NC, USA
2Department of Radiology, Duke University Medical Center, Durham, NC, USA
3Department of Pharmacology and Cancer Biology, Duke University Medical Center, Durham, NC, USA
Background: The overexpression of complement factor H (CFH) is associated with poor prognosis in lung cancers (1), and we have shown earlier that patients with early stage lung cancer had significantly higher incidence of anti-CFH antibodies than those with late-stage disease (2). A therapeutic antibody, GT103 was developed by isolating and cloning antigen specific B cells from patients expressing autoantibodies to complement factor H. GT103 was shown promote complement activation, kill tumor cells in vitro by complement CDC and inhibits tumor growth in vivo (3,4). We report here that GT103 also exerts an immune modulatory effect within the tumor micro-environment.
Material and methods: Tumor growth studies were done using the syngeneic CMT167 mouse lung cancer model. Immune profiling of tumors from IgG2 control and GT103-treated mice was done using flow cytometry. Genome wide analysis of tumors was performed using bulk and single cell RNA sequencing.
Results: Genome wide analysis and B cell depletion studies suggest that GT103 activates B-cell receptor signaling. Further, ScRNA-seq demonstrated that not only total intratumoral CD4+, CD8+ T and B lymphocytes were elevated in GT103-treated mice but Tregs, monocytic MDSCs and neutrophils with an immunosuppressive profile were reduced in GT103-treated tumors.
Discussion: In addition to increasing the influx and activation of immune stimulatory T and B cells, GT103 also limits immune suppressive cell populations like Tregs, and monocytic MDSC and neutrophils in the tumor micro-environment.
Conclusion: GT103 treatment creates a favorable immune microenvironment and induces anti-tumor immunity thus limiting the growth of CMT167 lung tumors.
References
Cui T., Chen C., Knösel T., Yang L., Zöller K., Galler K., Berndt A., Mihlan M., Zipfel P.F., & Petersen I. (2010). Human complement factor H is a novel diagnostic marker for lung adenocarcinoma. International journal of oncology, 39: 161-168.
Amornsiripanitch, N., Hong, S., Campa, M. J., Frank, M. M., Gottlin, E. B., & Patz, E. F., Jr. (2010). Complement factor H autoantibodies are associated with early stage NSCLC. Clin Cancer Res, 16(12), 3226-3231.
Campa, M. J., Gottlin, E. B., Bushey, R. T., & Patz, E. F., Jr. (2015). Complement Factor H Antibodies from Lung Cancer Patients Induce Complement-Dependent Lysis of Tumor Cells, Suggesting a Novel Immunotherapeutic Strategy. Cancer Immunol Res, 3(12), 1325-1332.
Bushey RT, Moody MA, Nicely NL, Haynes BF, Alam SM, Keir ST, Bentley RC, Roy Choudhury K, Gottlin EB, Campa MJ, Liao HX, Patz EF Jr. (2016). A Therapeutic Antibody for Cancer, Derived from Single Human B Cells. Cell Rep, 15(7), 1505-1513.
t-SNE plot showing distribution of cells per cluster in IgG2_Ctrl vs GT103 treated groups. ScRNA seq was performed on TILs from CMT-167 tumors treated with GT103 or IgG2 control antibody. The anchor genes between samples, identified using the FindIntegrationAnchor function, were used as a basis for Seurat to perform integration of the samples. The data were scaled before running Principal Component Analysis (PCA). Cluster identification and annotation were performed using a shared nearest neighbor modularity optimization-based clustering algorithm and FindNeighbors function with 50 principal components, followed by the FindClusters function with the Louvain algorithm using a 0.5 resolution.
Childhood Onset C3 Glomerulopathy: Recurrence After Kidney Transplantation
¹Nephrology institute, Schneider children’s medical Center, Petah Tiva ISRAEL
²Sackler School of Medicine, Tel Aviv University, ISRAEL
³Department of Nephrology and Hypertension, Rabin Medical Center, Beilinson Campus, Petach Tikva
Background: C3 Glomerulopathy (C3G) has been has been re classified as a glomerular complement mediated disease, with predominant C3 deposits almost a decade ago. Mutations and risk haplotypes in several complement system components and circulating autoantibodies result the loss of control of the alternative pathway has been described in patients with C3G.i,ii
High incidence of disease recurrence after transplantation is reported – between 30 and 77%, including graft failure in 17-50% of cases. Modalities of preventing and treating C3G recurrence after kidney transplantation include – plasma exchange, Rithuximab, Eculizumab with various reports regarding success.iii,iv Disease recurrence rate after transplantation in childhood onset C3G is still unknown.
The aim of our study was to retrospectively describe our cohort of childhood onset C3G transplanted patients, including treatment modalities and outcomes.
Methods: Retrospective cohort study, data regarding patients diagnosed with C3G as children or adolescents and underwent kidney transplantation between 2015-2020 was collected, including complement workup, treatment modalities and outcomes.
Results: Five patients underwent kidney transplantation, in four (80%) disease recurrence was diagnosed. Two patients improved after Eculizumab treatment (both had factor H antibodies), one patient reached graft failure 9 months after transplantation despite treatment with Eculizumab, one patient showed histologic signs of disease recurrence without clinical signs.
Conclusions: C3G recurrence rate after kidney transplantation in patients diagnosed as children or adolescents may be higher than described in the literature, treatment was successful only in part of the patients, new treatments are needed in order to safely transplant C3G patients and avoid disease recurrence.
References
Riedl M, Thorner P, Licht C. C3 Glomerulopathy. Pediatr Nephrol. 2017 Jan;32(1):43-57. doi: 10.1007/s00467-015-3310-4. Epub 2016 Apr 7. PMID: 27056062..
Pickering MC, D’Agati VD, Nester CM, et al. C3 glomerulopathy: consensus report. Kidney Int. 2013 Dec;84(6):1079-89. doi: 10.1038/ki.2013.377. Epub 2013 Oct 30. PMID: 24172683; PMCID: PMC3842953.
Zand L, Lorenz EC, Cosio FG et al, Clinical findings, pathology, and outcomes of C3GN after kidney transplantation. J Am Soc Nephrol. 2014 May;25(5):1110-7. doi: 10.1681/ASN.2013070715. Epub 2013 Dec 19. PMID: 24357668; PMCID: PMC4005307.