
Abstract
Rationale:
Atypical hemolytic uremic syndrome, which has a high probability of chronic kidney disease, morbidity, and mortality, needs to be promptly recognized when patients present with microangiopathic hemolysis.
Presenting Concerns of the Patient:
Three patients present with laboratory parameters consistent with a thrombotic microangiopathy. With a suspected diagnosis of thrombotic thrombocytopenic purpura, steroids with plasmapheresis were initiated.
Diagnoses:
With ADAMTS13 levels reported normal, the suspected diagnoses were reevaluated. Given ongoing renal impairment, atypical hemolytic uremic syndrome was strongly considered.
Interventions:
When local funding issues precluded the prompt use of eculizumab, 4 doses of weekly rituximab were trialed.
Outcome:
Over 2 years later, all 3 patients have sustained durable remissions defined by the absence of kidney impairment or laboratory investigations concerning for microangiopathic hemolytic relapse.
Lessons Learned:
In cases of a suspected autoimmune mechanism leading to atypical hemolytic uremic syndrome, long-term use of eculizumab may not be required.
What was known before
The complement inhibitor eculizumab remains an effective treatment option for atypical hemolytic uremic syndrome. Multiple congenital mutations and autoimmune mechanisms have been described leading to the clinical manifestations. In pediatric cases of autoantibodies to complement factor H, an international consensus has recommended considering cessation of eculizumab when the antibody titer is <1000 AU/mL.
What this adds
The high cost of eculizumab has limited its prompt availability in some health authorities. After successfully treating 3 adult patients presenting with suspected atypical hemolytic uremic syndrome with only steroids, plasmapheresis, and rituximab, the strategy for long-term remission should be further investigated.
Introduction
More readily available testing of von Willebrand factor–cleaving protease levels (ADAMTS 13) to accurately diagnose thrombotic thrombocytopenic purpura (TTP) has led to earlier recognition of atypical hemolytic uremic syndrome (aHUS), a much rarer microangiopathy. With higher incidence of chronic kidney disease, morbidity, mortality, and vastly differing pathophysiology, treatment strategies are tailored in effort to improve prognosis.1,2 Approved in 2011, the terminal complement inhibitor eculizumab is an effective option. 1 A barrier remains the high cost of the drug with some recommendations for indefinite treatment in the setting of persistent compromised kidney function. Other strategies have included kidney–liver transplant in cases with defined genetic mutations of complement regulation, or immunosuppression in those with an autoimmune component.1-11 We describe a case series of successful treatment using plasmapheresis, steroids, and rituximab in 3 adult patients presenting with microangiopathic hemolytic anemia (MAHA).
Case 1
A 51-year-old female, with a medical history of migraine and hypothyroidism, presented with a week-long history of fatigue, fever, jaundice, bruising, paresthesia in distal extremities, and intermittent blurred vision with headache. Initial blood work discovered microangiopathic hemolysis with a hemoglobin of 56 g/L, platelets 30 × 109/L, and a white blood cell (WBC) count of 6.8 × 109/L. Hemolytic markers were consistent with hemolysis, and the peripheral smear showed increased polychromasia with both spherocytes and schistocytes. Renal function was compromised with an estimated glomerular filtration rate (eGFR) of 26 mL/min/1.73 m2. There was no history of recent diarrheal illnesses or culprit medications. Red blood cells were transfused, steroids administered (intravenous [IV] solumedrol followed by daily prednisone 1 mg/kg dosing), and plasmapheresis initiated for a presumed diagnosis of idiopathic TTP. The platelet count began to increase after 2 days of treatment, normalized by day 10, and by day 14, there was normalization of the bilirubin and lactate dehydrogenase (LDH) (Figure 1). Despite the reassuring blood cell counts (CBC), and resolution of hemolysis markers, renal function worsened slightly over the first week before the eGFR stabilized consistently between 30 and 41 mL/min/1.73 m2 (creatinine 130-150 μmol/L) over the subsequent 3 weeks. Sixteen days into the hospital admission, pretreatment ADAMTS13 level was reported at 103%. Financial coverage for eculizumab was submitted, daily prednisone and plasmapheresis continued, and rituximab (375 mg/m2 weekly × 4) was trialed. Creatinine normalized 12 days after rituximab was started. The pretreatment factor H autoantibody was reported at 64 U/mL (normal <22 U/mL). Six months off all treatment, classical and alternative complement function testing remained normal including testing for C3, C3d, C5b-9, and factor I levels, and there were no detectable anti–factor H antibodies. Over 3 years later, blood counts, hemolytic markers, and kidney function remain normal. Funding for eculizumab was denied but would be reconsidered in relapse. Genetic testing did not find any culprit mutations.

Case 1 platelets and GFR.
Case 2
A 50-year-old male presented to an emergency room while traveling with nonbloody diarrhea. He was treated with metronidazole without a stool culture being collected. Two weeks later, he again presented to his local emergency room with petechiae and bruising; lab work revealed a hemoglobin of 130 g/L, platelets 14 × 109/L, WBC 7.5 × 109/L, and a eGFR of 36 mL/min/1.73 m2. With peripheral blood film reporting schistocytes, plasmapheresis and high-dose steroids (prednisone 1 mg/kg daily) were initiated for a suspected diagnosis of idiopathic TTP. On day 8 of treatment, ADAMTS13 level (preplasmapheresis) was reported at 81% (Normal 0.40-1.30 U/mL). As daily plasmapheresis continued, the platelet count and creatinine were slow to respond. Malignancy investigations including a computed tomography (CT) chest/abdomen/pelvis and a bone marrow biopsy were negative. With no preplasmapheresis plasma sample available to send for aHUS testing, and local funding issues precluding the use of eculizumab, anti–factor H autoantibodies were considered a strong possibility. Rituximab 375 mg/m2 weekly × 4 was started on day 12; plasmapheresis continued prior to each infusion and held the day after. Platelets normalized by day 16, and creatinine normalized by day 43 (Figure 2). Complement activation testing done 2 months after finishing rituximab revealed normal levels of classical and alternative pathway complement function. Two years after initial presentation, there is no evidence of relapse with persistently normal blood cell counts, chemistries, and renal function.

Case 2 platelets and GFR.
Case 3
A 27-year-old male, with a medical history of pancreatitis presented with abdominal pains and a recent history of alcohol consumption and cocaine use. Blood work revealed a hemoglobin of 106 g/L, platelets 8 × 109/L, and a WBC count of 8.6 × 109/L. Peripheral smear reported findings of MAHA with spherocytes and 3% schistocytes. Renal function was compromised with a eGFR of 15 mL/min/1.73 m2 (creatinine of 437 μmol/L, normal 1 day prior) and lipase elevated at 1609 U/L. With no history of a diarrheal illness and ongoing rapid hemolysis, packed red blood cells were transfused, IV solumedrol administered, and plasmapheresis initiated for suspected idiopathic TTP. One day later, the ADAMTS13 level was reported at 59%, aHUS testing ensued. Positive anti-PR3 and p-ANCA antibodies were also found suggesting the possibility of vasculitis or levamisole exposure. The platelet count began to climb 5 days into treatment, but renal function remained poor requiring initiation of dialysis 15 days into hospital admission (creatinine consistently >700 μmol/L). As funding for eculizumab was not approved, steroids and plasmapheresis continued, and rituximab (375 mg/m2 weekly × 4) was trialed. Just prior to initiation of the second rituximab dose, pre-plasmapheresis aHUS testing was reported showing an increase in both the classical and alternative complement pathway activity (164% and 169%, respectively), and an increased level of C5b-9 at 1048 ng/mL (normal <320 ng/mL). Testing of C3, C3d, factor H, and factor I levels were normal, and there were no reported anti–factor H antibodies. With a now documented improvement in kidney function, only requiring 4 total dialysis treatments, despite no antibodies found, rituximab continued weekly for 4 weeks (Figure 3). Plasmapheresis was discontinued after the third dose of rituximab with the platelets now normal, and eGFR above 30 mL/min/1.73 m2. Two years later, blood cell counts and renal function remain normal.
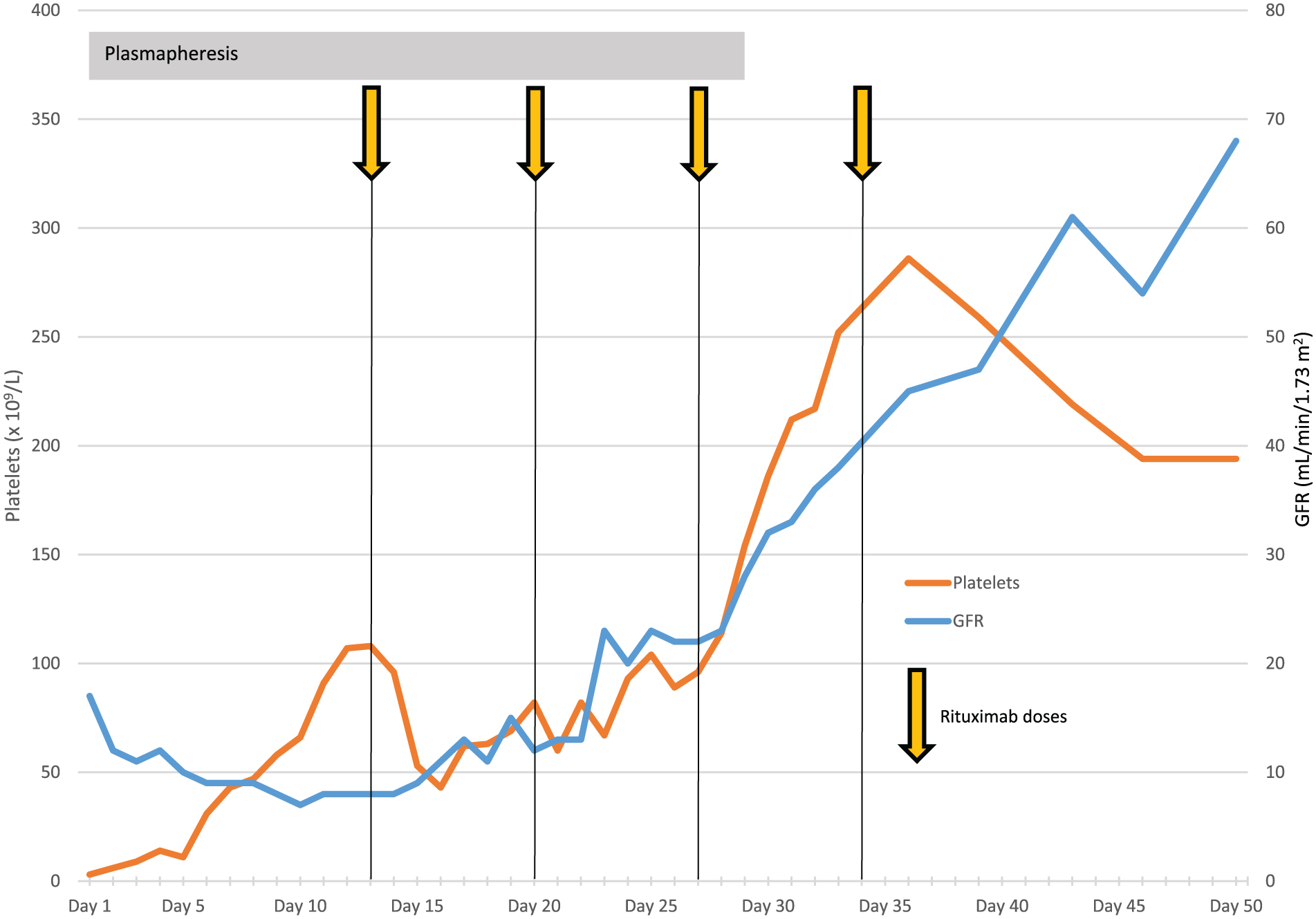
Case 3 platelets and GFR.
Discussion
Classically described as the thrombotic microangiopathies (TMA), TTP (congenital and acquired), shiga toxin–associated HUS, aHUS, drug-induced TMA, disseminated intravascular coagulopathy, HELLP syndrome, scleroderma renal crisis and malignant hypertension, among others, present with similar laboratory presentations. In TTP, tissue injury results from large von Willebrand factor multimers aggregating platelets with subsequent microvascular thrombosis and ischemia.2,3 In contrast, aHUS results from uncontrolled activation of the complement system with subsequent increased anaphylatoxin production and membrane attack complexes.2,3 This cytotoxicity to the endothelial cells leads to increased swelling and subendothelial expansion exposing the prothrombotic components to the vascular space. The subsequent increased vascular permeability and activation of the coagulation cascade with fibrin contributes to increasing edema in vital organs and the microangiopathy. 2 The clinical presentations of aHUS can include mental status changes or seizures from brain edema, cardiac dysfunction or pericardial effusions, pulmonary infiltrates or pleural effusion, pancreatitis, ascites, advanced renal failure, or even anasarca. The vessel wall is thought to remain relatively intact in TTP, thus not resulting in the same complications of increased vascular permeability. 2 These clinical complications should therefore not be seen to the same degree in acquired TTP in the absence of other comorbidities.
Many mutations have been identified leading to dysregulation in the complement system; most common are loss of function of complement regulatory proteins (complement factor H or I, membrane cofactor protein, or thrombomodulin).2,3 Also described are gain of function mutations (C3, complement factor B) resulting in less degradation of critical convertases needed for the propagation of the cascade. Most infrequently (5%-10% of cases) autoantibodies to complement factor H (CFH-Ab) result in decreased ability to suppress the process.2,3 Although more commonly seen in the pediatric population, cohort studies have shown the CFH-Ab mechanism is more frequently seen in later childhood therefore certainly needs to be considered in adults.4,5 In this subset of patients, the mechanism is postulated to involve an acquired factor in the setting of genetic predisposition as >90% of patients with CFH-Ab also have a homozygous gene deletion resulting in complete deficiency of complement factor H–related 1 and 3 proteins (CFHR1 and CFHR3).4-9 In terms of clinical presentation, when compared with other complement mutation–associated HUS, increased extrarenal complications are described including more seizures, pancreatitis, hepatitis, and cardiac symptoms. 5 When compared with aHUS as the result of factor H mutations, antibody-associated diseases have lower frequencies of end-stage renal disease and death. 5
With ADAMTS13 levels becoming more readily available and standardized, a diagnosis of aHUS needs to be strongly considered if a level >10% is found in the setting of a microangiopathic clinical presentation. This is especially true with significant concurrent renal compromise in the absence of Shiga toxin–related diarrhea. Plasmapheresis remains first-line treatment of MAHA with no history of a recent diarrheal illness, known metastatic malignancy, or offending medications. The procedure acutely replaces both ADAMTS13 and defective complement regulatory proteins. Response in aHUS is dependent on the culprit mutation; those with inactivation of plasma proteins or gain of function mutations predicted to respond better than mutations in the membrane-bound proteins. 2 In cases with autoantibodies, plasmapheresis reduces the antibody titer, but the best strategy for long-term remission remains unclear. Patients with aHUS treated with only plasmapheresis and no eculizumab have a significantly higher mortality rate, reported at 20% to 30%, as well as more persistent renal compromise compared with patients with TTP. 2
If aHUS is recognized early and treated with eculizumab, prospective data report a >85% recovery as evidenced by improvements in platelet count and renal status, and normalization of hemolytic parameters. 1 Neither eculizumab nor plasmapheresis, however, directly addresses an underlying autoimmunity. Rituximab has an ever-expanding role in TTP with high rates of remission reported (70%-90%) in those patients with resistant disease. 2 It has also been used in patients with continued low ADAMTS13 levels in remission to prevent relapse. For aHUS with demonstration of CFH antibodies, case reports have been published using various immunosuppressive combinations including prednisone, azathioprine, mycophenolate mofetil, cyclophosphamide, and rituximab.7-9
The first published adult case using only plasmapheresis, corticosteroids, and rituximab in the treatment of aHUS with CFH-Ab (32 000 UI/L) was in a 42-year-old male. 8 Despite a documented reduction in CFH-Ab with steroids and plasmapheresis, the patient remained dependent on hemodialysis until rituximab was given. A second case (anti-CFH antibodies at 82 000 AU/mL) also documented hematological and renal remission achieved at day 31 post rituximab initiation, negative antibodies at day 45, and free from relapse over 3 years later. 9 In a third case, rituximab was trialed after no renal function recovery was seen after 2 doses of eculizumab. 10 After 4 doses of eculizumab and 3 doses of rituximab, renal function did improve, but 3 months after the complement inhibitor was stopped, the TMA resurfaced. This patient, however, was also discovered to have CFH and CD46 genetic abnormalities predisposing aHUS with no anti-CFH antibodies reported. Favorable outcomes with CFH autoantibodies were also reported in 4 pediatric cases using cyclophosphamide pulses, steroids, and plasmapheresis with up to 6 years of sustained remission. 7 In 1 patient, rituximab with plasmapheresis alone was used unsuccessfully in preventing relapse, with CFH antibody titers increasing after only 1 month. A reduction of antibody titer has been demonstrated to correlate with remission. In most patients, however, the antibody remains detectable and there are insufficient data to determine a threshold above which relapse is imminent.5,7 A cohort review in 138 children demonstrated independent risk factors for adverse outcome include an antibody titer >8000 AU/mL, a low C3 level, and a delay in plasmapheresis ≥17 days from time of onset. 4 This has led to the recommendation by an international consensus of cessation of eculizumab in children when the anti-CFH antibody titer is <1000 AU/mL.10,11 Other cases have been reported involving the use of rituximab in preparation for renal transplant, with varying results regarding the continued presence of the antibody. 5 In these cases, continued use of immunosuppression for graft rejection may also be key in keeping the aHUS immune process under control.
Much still needs to be learned regarding the pathophysiology, diagnosis, and treatment for atypical HUS and TMAs in general. As duration of eculizumab treatment in aHUS remains in question, with relapse rates off treatment reported at 20% to 31%, a cessation trial is reasonable once organ function improves and, if present, a precipitant illness improves. 12 A comparison of the clinical and laboratory presentations of the 3 cases is illustrated in Table 1. With no preplasmapheresis sample available for specific aHUS testing in case 2, and the history of cocaine use with positive anti-PR3 and p-ANCA antibodies in case 3 (suggesting the possibility of vasculitis or levamisole exposure with a TMA presentation), the authors recognize the diagnoses are not definitive. In cases with an anti-CFH antibody present, plasmapheresis and/or eculizumab continue to be recommended for acute treatment. The best strategy for long-term remission remains in question. Despite only finding a positive antibody in one of our 3 cases, the microangiopathic process was reversed and dramatic improvements in kidney function were seen in all 3 patients. More research needs to be done regarding better targeted therapies. This case series would suggest a potential benefit to using rituximab in patients with aHUS, especially with the possibility of alternate autoimmune mechanisms beyond anti-CFH antibodies yet to be discovered.
Comparison of the Clinical and Laboratory Patient Presentations.

Note. eGFR = estimated glomerular filtration rate; CFH = complement factor H; aHUS = atypical hemolytic uremic syndrome; LD, Lactate Dehydrogenase.
Footnotes
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Alexion supports local laboratory testing for ADAMTS13 with an unrestricted grant.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics Approval and Consent to Participate
Ethics approval is not needed for a case report at Dr. Patterson’s institution.
Consent for Publication
All patients provided written informed consent to publish their cases.
Availability of Data and Materials
Not applicable