
Abstract
Rationale:
Geller et al reported a rare mutation in the mineralocorticoid receptor (MR) resulting in constitutive MR activity. Progesterone, normally an MR antagonist, acts as a potent agonist with this mutation. Progesterone levels can increase 100-fold during pregnancy and thus lead to increased MR activity in this setting, resulting in hypertension (HTN) and hypokalemia during pregnancy and resolution of hypokalemia after delivery.
Presenting concerns:
Our patient was a 33-year-old African American female with a history of pregnancy-induced HTN associated with hypokalemia during her last pregnancy. She presented with muscle weakness from profound hypokalemia complicated by nephrogenic diabetes insipidus (DI) and rhabdomyolysis.
Diagnosis:
Her admission potassium was 1.9 mmol/L (3.5-5.1 mmol/L) with a 24-hour urine potassium of 35 mmol per day and an unmeasurable serum aldosterone level. Her potassium normalized 1 day after delivery off potassium supplementation and amiloride, which were last given 1 day prior to her delivery. Recurrent hypokalemia from nonaldosterone-mediated renal potassium wasting during pregnancy (with normal potassium in a nongestational state) is consistent with the cases of gain-of-function mutation in MR that Geller et al report. A definite diagnosis requires genetic analysis.
Interventions:
Her hypokalemia was refractory to potassium replacement but quickly responded to an inhibitor of the epithelial sodium channel (ENaC), amiloride.
Outcomes:
Her potassium normalized on amiloride 10 mg per day and KCL 40 mEq daily during the remainder of her pregnancy, and her nephrogenic DI resolved after this correction of hypokalemia. After her delivery, her potassium remained normal off the potassium supplements and amiloride.
Novel findings:
Pregnancy-induced hypokalemia from an activating MR mutation has rarely been reported. Pregnancy-induced HTN is often the first differential diagnosis in a patient who develops worsening in her HTN during pregnancy. We should also consider the possibility of a gain-of-function mutation in MR in these patients who also have associated hypokalemia.
Introduction
We report a patient with a history of pregnancy-induced hypertension (HTN) who experienced hypokalemia during her last pregnancy.
Presenting Concerns
The patient was a 33-year-old African American female G6P5005 who had a history of pregnancy-induced HTN since her first pregnancy when she was 16 years old. She developed persistent HTN after her fourth pregnancy in 2013. She developed hypokalemia during her fifth pregnancy in 2018, with serum potassium ranging from 2.9 to 3.1 mmol/L (3.5-5.1 mmol/L). Her blood pressure before the Cesarean section in 2018 was 189/93 and went down to 137/84 after the surgery. She had not been following with her primary care physician, and her potassium as well as her blood pressure were not checked after that pregnancy. Furthermore, her potassium level was not checked during her fourth pregnancy in 2013 at our hospital, and there was no information on the potassium level during her first 3 pregnancies. She had been taking labetalol 400 mg twice daily for her HTN over the past several years, and nifedipine was added during this pregnancy. She presented to our institution at 35-weeks pregnant with a 1-week history of generalized weakness with difficulty ambulating. She had also developed polydipsia and polyuria several days prior to admission. The patient denied having any diarrhea, nausea, vomiting, palpitations, diaphoresis, or anorexia, or any history of pica, surreptitious diuretics, or laxative use. She had been taking an elderberry supplement (without Licorice) once a day for years. Her family history was significant for HTN in her mother, but the patient was not aware of any hypokalemia in her family. She experienced frequent headaches, which usually worsened during pregnancy, and had used 90 tablets of Butalbital-acetaminophen-caffeine within 2 to 3 weeks for her headaches prior to admission.
Clinical Findings
Her physical exam revealed a respiratory rate of 20, uncontrolled blood pressure of 182/86 without peripheral edema, and no signs suggestive of Cushing’s syndrome or abnormal sexual development. Her initial potassium was 1.9 mmol/L, and the obstetrics service arranged for potassium replacement. Her serum bicarbonate was 22 mmol/L (21-30 mmol/L) and magnesium 2.2 mg/dL (1.6-2.6 mg/dL).
Timeline
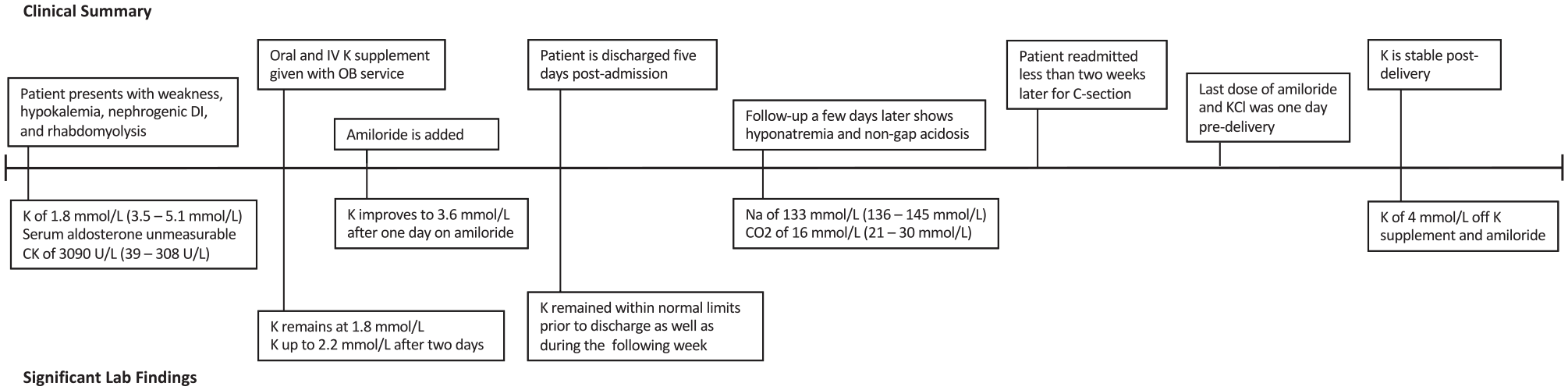
Diagnosis Focus and Assessment
The initial impression from her obstetrician was Butalbital-acetaminophen-caffeine-induced hypokalemia. However, she did not exhibit typical clinical constellations of Butalbital-acetaminophen-caffeine toxicities such as hypotension, nausea, and vomiting. The Butalbital-acetaminophen-caffeine dosage that she used (on average every 4 hours) was at the upper limit of the recommended dose. Furthermore, her caffeine level was normal at 3 µg/mL (≤ 20 ug/mL). A reported case of Butalbital-acetaminophen-caffeine-induced hypokalemia occurred in a patient with suspected massive overdose and was thought to be related to caffeine-induced diuresis. 1 A 24-hour urine potassium, serum renin, and aldosterone were sent by her obstetrician on the day of her admission. A total of 120 mEq of potassium chloride (KCL) (60 mEq intravenous and 60 mEq oral) were given by obstetrics service without any significant improvement in her potassium level (potassium of 1.8 mmol/L the following day), and a nephrology consultation was requested. Table I in the supplementary material highlights some of her lab findings; findings were significant for aldosterone < 3 ng/dL, renin 0.5 ng/mL/hour (supine 0.2-1.6 ng/mL/hour; upright 0.5-4 ng/mL/hour), and 24-hour urine potassium of 35 mmol/TV. The urine potassium of 35 mmol per day in the setting of hypokalemia is consistent with renal potassium wasting. The trans-tubular potassium gradient (TTKG) value of 5 in the setting of hypokalemia is also supportive of renal potassium loss (with the limitation in interpreting the value from low urine sodium and osmolality).
Therapeutic Focus and Assessment
Her potassium only slightly improved to 2.2 mmol/L 2 days into her admission despite ongoing KCL replacement. Given the possibility of a gain-of-function MR mutation, amiloride 10 mg per day, a pregnancy category B agent, was added that morning with improvement of potassium to 3 mmol/L in the evening and 3.6 mmol/L the following day. DDAVP 1 mcg IV was given for a few doses that day as well when she had 16 L of urine per day without significant changes in urine output. Her urine output was about 7200 mL/day on her second day of admission, 16 100 mL/day on the third day of admission, 9000 mL/day 3 days after that, and 3000 mL/day 2 days after that (4 days after correction of hypokalemia). Her potassium remained normal around 4.5 to 5 mmol/L on amiloride 10 mg daily and KCL 40 mEq daily after her discharge. Her follow-up outpatient’s lab 2 days postdischarge showed hyponatremia and nongap acidosis with a sodium of 133 mmol/L (136-145 mmol/L), anion gap of 14 mmol/L (5.0-14.0 mmol/L), and CO2 of 16 mmol/L (21-30 mmol/L). Her serum albumin level was 3.8 g/dL (3.97-4.97 g/dL). The development of hyponatremia could be related to a side effect of amiloride or an unmasking of her pregnancy-associated hyponatremia after improvement in nephrogenic DI. Low serum bicarbonate similarly could be secondary to amiloride or an unmasking of a metabolic compensation in pregnancy-induced respiratory alkalosis after correction of pseudo-hyperaldosteronism with an ENaC blocker.
Follow-Up and Outcomes
Her amiloride and KCL were not given when she was readmitted for her regularly scheduled Cesarean section almost a week later. She took her last dose of amiloride and KCL in the morning 1 day prior to her delivery. She was maintained on nifedipine 60 mg daily and labetalol 400 mg twice daily with an acceptable blood pressure after the delivery. Her potassium 1 day postdelivery was normal at 4 mmol/L. She did a telehealth follow-up a few days later and reported a blood pressure of 116/65 on a lower dose of nifedipine at 30 mg daily and the same dose of labetalol. A follow-up chemistry panel, urine electrolytes, renin, aldosterone, and 24-hour urine for free cortisol and cortisone were ordered but not completed despite our recommendation.
Discussion
In normal pregnancy, there are alterations in fluid, electrolytes, renin-angiotensin-aldosterone system (RAS), and various gestational hormones. Hyponatremia and respiratory alkalosis are commonly encountered during pregnancy. 2 Aldosterone levels are elevated with concentration as high as 10-fold increase compared with nonpregnant states. In preeclampsia, aldosterone levels are significantly lower than normal pregnancy approaching pregestational level. 3 In pregnancy, there are several physiologic changes that can lead to hypokalemia specific to pregnancy. For example, hyperemesis gravidarum can lead to hypokalemia from gastrointestinal loss. In 2 pregnant patients with aldosterone-producing adenomas with activating CTNNB1 mutation with increased luteinizing hormone-chorionic gonadotropin and gonadotropin-releasing hormone encoding gonadal receptors, these patients developed HTN and hypokalemia during their pregnancies. 4
Primary hyperaldosteronism (PA), often an underrecognized cause of HTN in pregnancy, needs to be considered in pregnant patients with severe HTN with or without hypokalemia. Elevated progesterone level during pregnancy competitively inhibits the action of aldosterone at the MR receptors; therefore, hypokalemia might not be present until postpartum. Diagnosis of PA is typically made in patients who have an elevated aldosterone-renin-ratio (ARR). This ratio can be falsely negative during pregnancy since renin production in pregnancy is relatively higher than aldosterone production resulting in a lowered ARR. 5 Confirmatory tests of PA such as the saline loading test and captopril challenge test are contraindicated during pregnancy. 6 Therefore, diagnosis of PA during pregnancy can be challenging.
Pseudo-hyperaldosteronism mimics the clinical effects of elevated aldosterone such as HTN, hypokalemia, and metabolic alkalosis. The differential diagnoses in a patient with HTN and hypokalemia associated with urinary potassium wasting without elevated serum aldosterone include ectopic ACTH production, Cushing syndrome, Liddle syndrome, apparent mineralocorticoid excess (AME) syndrome (which might be hereditary or acquired from Licorice use or azole antifungals use, such as with itraconazole or posaconazole), 11-beta hydroxylase deficiency (usually associated with virilization), 17-alpha hydroxylase deficiency (typically associated with abnormal sexual development), and an activating MR mutation.7,8 In our case, her urinary potassium excretion of 35 mmol per day in the setting of profound hypokalemia is consistent with renal potassium loss as the cause of her hypokalemia. Her serum aldosterone was unmeasurable. She lacked clinical features of Cushing syndrome with a fasting glucose of 81 mg/dL (74-106 mg/dL) on admission, normal ACTH, and normal cortisol albeit slightly elevated 24-hour urine cortisol but below the 3-fold increase threshold for diagnosis of Cushing disease. These findings are not supportive of ectopic ACTH and Cushing disease. She did not take any Licorice supplement. The hereditary form of AME syndrome is usually a disease of childhood. She does not have any abnormal sexual development to suggest 11-beta hydroxylase or 17-alpha hydroxylase deficiency. Patients with Liddle syndrome should have hypokalemia in a nongestational state as well. The normalization of potassium after delivery without amiloride or potassium supplement leads us to hypothesize that her recurrent pregnancy-induced hypokalemia can be explained by an MR mutation, as Geller et al described. Other causes of pseudo-hyperaldosteronism cannot be completely excluded with the lack of complete information about her potassium outside her pregnancy and peri-partum period due to non-compliance issues in follow-up. A definite diagnosis requires a genetic study.
Her hypokalemia was resistant to potassium supplement but was rapidly responsive to amiloride which inhibits ENaC and thus indirectly ROMK activity. The development of hypokalemia-induced nephrogenic DI with her urine output as high as 16 L per day can worsen the degree of hypokalemia. Increased luminal flow in cortical collecting tubules is known to cause increased intracellular calcium concentration which leads to activation of flow-dependent Maxi-K channels. 9 A brain MRI showed mild flattening of the pituitary without expansion of the sella turcica and without pituitary enlargement or macroadenoma. This suggested a partially empty sella turica (PES) which might be responsible for her headaches. Although PES can cause abnormal anterior and more infrequently posterior pituitary hormone secretion, 10 her polyuria did not respond to DDAVP and spontaneously resolved a few days after correction of hypokalemia. This is supportive of hypokalemia-mediated nephrogenic DI rather than central DI.
She had a history of pregnancy-induced HTN since her first pregnancy, albeit no information on her potassium concentrations was available before her fifth pregnancy. It is conceivable that her potassium concentration might not have been checked like it was not during her fourth pregnancy or her hypokalemia was simply treated without follow-up. Geller et al 11 described a gain-of-function mutation in MR, specifically a missense mutation resulting in substitution of leucine for serine at codon 810. Carriers for this mutation showed marked increase in HTN as well as suppression of aldosterone secretion. There was no significant hypokalemia among the carriers in a normal physiologic state. Twenty-one-carbon steroids, such as progesterone which normally acts as an antagonist to wild-type MR, activate MR in patients with this mutation. Interestingly, spironolactone, which normally inhibits wild-type MR, activates the mutant MR. In the report by Geller et al, 2 carriers with MR mutations developed exacerbation of HTN and hypokalemia during all their pregnancies. One carrier required urgent Cesarean section at 34-weeks’ gestation due to worsening in blood pressure (210/120 mm Hg). The other carrier had exacerbation of HTN requiring early delivery in the sixth and seventh months of gestation. Neither carrier had proteinuria or edema. In another report by Garg et al, 7 a 23-year-old G2P1A0 developed hypokalemia during her first and second pregnancies with normal potassium after delivery. In this report, the patient’s blood pressure was 138/81 mm Hg when she was at 32-weeks of gestation. She had induction of labor at 37-weeks of gestation because of uncontrolled HTN and hypokalemia, but the exact blood pressure level was not specified.
In our case, the initial metabolic panels did not show evidence of an acid-base disorder. Metabolic alkalosis, which is a feature of hyperaldosteronism, was not mentioned in the report by Geller et al and was not present in the report by Garg et al.7,11 Metabolic compensation for chronic respiratory alkalosis which is common in pregnancy might mask the usual clues for detecting metabolic alkalosis. Similarly, in our case, with her normal bicarbonate level, blood gas was not performed. In retrospect, her urine pH was at the upper limit of normal at 8 (5-8), which might indicate renal compensation for respiratory alkalosis. The development of low serum bicarbonate several days after amiloride treatment might reflect a metabolic compensation of respiratory alkalosis that was unmasked after resolution of her hypokalemic metabolic alkalosis. Aldosterone activates Na+/H+ exchange, and hyperaldosteronism causes urinary acidification resulting in metabolic alkalosis from excessive urinary chloride loss.12,13,14 In our case, urine chloride was low at <20 mmol/dL. In the setting of respiratory alkalosis, the renal net acid secretion decreases largely from bicarbonaturia and decreases in the excretion of titratable acid. 15 Urine pH can be alkaline in the setting of hyperaldosteronism if distal urinary acidification is impaired. 12 We postulate that the concomitant respiratory alkalosis with renal compensation might explain the apparently normal acid-base values from the chemistry panel, alkaline urine, and low urinary chloride in our patient.
Conclusion
Pregnancy-induced HTN is often the leading differential diagnosis of worsening in HTN during pregnancy. This case illustrates the need to consider an activating MR mutation as a cause of exacerbation of HTN during pregnancy in the setting of hypokalemia that is secondary to nonaldosterone-mediated renal potassium wasting. Amiloride, a pregnancy category B agent, proved effective in both potassium and blood pressure control in our case.
Supplemental Material
sj-pdf-1-cjk-10.1177_20543581211017424 – Supplemental material for A Case Report of Recurrent Hypokalemia During Pregnancies Associated With Nonaldosterone-Mediated Renal Potassium Loss
Supplemental material, sj-pdf-1-cjk-10.1177_20543581211017424 for A Case Report of Recurrent Hypokalemia During Pregnancies Associated With Nonaldosterone-Mediated Renal Potassium Loss by Pairach Pintavorn and Stephanie Munie in Canadian Journal of Kidney Health and Disease
Footnotes
Ethics Approval and Consent to Participate
The patient gave verbal consent to publish her information in de-identified form.
Consent for Publication
All authors reviewed this manuscript and provided consent for publication.
Availability of Data and Materials
Data are available from the authors upon reasonable request, according to the terms of relevant privacy legislation and regulation.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.