
Abstract
Background:
Urate nephropathy is a rare cause of acute kidney injury. Although most risk factors are associated with chemotherapy, tumor lysis syndrome or rhabdomyolysis, occurrence following severe seizure has also been reported. Uric acid measurement following convulsion is rarely performed and, therefore, the incidence of hyperuricemia in this context is unknown.
Objective:
The objective is to present a case of urate nephropathy following generalized tonic-clonic seizure (GTCS) and to investigate the kinetics of serum uric acid and creatinine levels in a series of patients admitted for severe seizures.
Design:
Retrospective case report and prospective case series.
Setting:
Emergency room department and neurology unit of a tertiary care hospital.
Patients:
The study included 13 hospitalized patients for severe GTCS.
Measurements:
Type, timing, and duration of seizure episodes were documented. Demographic data, weight, hypouricemic therapy, and baseline serum creatinine were recorded. Blood samples (uric acid, creatinine, blood gas, lactate, and creatinine kinase) and urine samples (uric acid, creatinine, and dipstick) were prospectively collected at Day 0, 1, 2, and 3 following the GTCS episode.
Methods:
We identified and described one rare case of urate nephropathy following GTCS. Then, we presented the kinetic of uric acid and creatinine levels and the acute kidney injury incidence over the follow-up period. All analyses were using descriptive statistics.
Results:
During the study period, 13 patients with a median tonic-clonic seizure duration of 5.0 minutes (interquartile range [IQR], 2.0–12.5) were included. From day 0 to day 3, the median serum uric acid level decreased from 346.0 µmol/L (IQR, 155.0–377.5) to 178.0 µmol/L (IQR, 140.0–297.5) and median serum creatinine from 73.0 µmol/L (IQR, 51.0–80.0) to 57.0 µmol/L (IQR, 44.0–70.0). Acute kidney injury occurred in four patients.
Limitations:
This is a single-center observational study with small sample size, which does not allow us to demonstrate causality between the increase of uric acid levels observed and the occurrence of acute kidney injury. A delay between the first sampling and seizure episodes was observed and could explain the limited increase of uric acid levels captured.
Conclusions:
There is a signal for an acute increase of uric acid levels following a severe seizure before returning to baseline within 3 days. During that period, there might be an increased risk of acute kidney injury, although these changes seem to be usually mild and reversible. Our findings suggest that routine serum uric acid measurement in patients presenting with GTCS could help to identify those patients at risk of developing acute kidney injury as a result of acute hyperuricemia. Further larger studies are required to confirm the effectiveness of such screening in acute kidney injury prevention.
Trial Registration:
As an observational noninterventional study, no registration was required.
Introduction
Seizures lead to acute metabolic changes depending on their type, length, and intensity. Numerous laboratory and metabolic abnormalities have been associated with severe convulsions,1-3 mimicking what is found in the context of intense physical activity.3,4 Previous studies in emergency rooms (ERs) showed that postictal lactic acidosis, ammonia, creatine kinase (CK), and prolactin elevations are relatively common.5,6 However, other less-known metabolic disturbances including electrolytes, uric acid, and osmolality changes have also been described following convulsions.3,7 Severe acute increases of serum uric acid enough to explain uric acid nephropathy have been rarely reported.8-11 Acute uric acid nephropathy should be suspected in high-risk patients who develop acute kidney injury (AKI) with significantly elevated serum uric acid and the presence of copious uric acid crystals in the urine sediment.10,12 Forty years ago, Kelton et al first proposed that a urine urate-to-creatinine ratio of more than 1.0 is highly suggestive of acute uric acid nephropathy. 13 As hyperuricemia can be effectively treated using urine alkalinisation and different pharmacologic agents, quickly identifying these patients at risk remains a major limitation. However, seizure-induced hyperuricemia is a rarely described complication, and even more its association with the occurrence of AKI.
Following a suspected case of seizure-induced urate nephropathy, we became interested in documenting and quantifying this specific and manageable metabolic disturbance following generalized tonic-clonic seizure (GTCS). In this study, the objective was to investigate prospectively this under recognized phenomenon and to describe the kinetics of uric acid and serum creatinine (SCr) in a series of patients admitted for severe GTCS.
Case
The patient was a 26-year-old Caucasian man of 74 kg with a past medical history of epilepsy with low convulsive threshold since the age of 19 and viral meningitis in childhood. His medications included levetiracetam, lamotrigine, and lacosamide. Two years earlier, an AKI episode following a GTCS was treated in another hospital. At that time, SCr reached 252 μmol/L without significant CK elevation. The uric acid level was not measured, and the SCr returned to baseline within 3 days with no clear etiology. At that time, SCr after discharge was 94 µmol/L (eGFR 97 mL/min/1.73 m2) with normal urine dipstick.
More recently, the patient was transported to the ER following a 2-minute witnessed GTCS. In the ER, second and third GTCS episodes of respectively 2- and 1-minute duration occurred, with incomplete return to baseline neurological state between episodes. His blood pressure was 123/68 mmHg and his oral temperature was 35°C. On physical examination, the patient had no sign of meningism or trauma. The cerebral scan was within normal limits. The initial laboratory workup revealed the following: severe lactate acidosis at 22.8 mmol/L (reference range: <2.4 mmol/L) with serum bicarbonate at 5.1 mmol/L (reference range: 21.0-28.0 mmol/L), elevated SCr at 134 µmol/L, serum electrolytes within normal limits and leukocytosis at 33 × 109/L (reference range: 4.0-11 × 109/L). The patient’s hemodynamic and vital parameters remained stable throughout the hospitalization. A lumbar puncture demonstrated the absence of white blood cells in the cerebrospinal fluid and bacterial culture and polymerase chain reaction (PCR) for all common viral infection of the central nervous system were negative. He was admitted to the neurology unit following a 24-hour surveillance at the ER, with a diagnosis of a resolved status epilepticus secondary to limited drug compliance and acute lack of sleep.
The SCr reached 213 µmol/L the following day and a nephrology consultation was requested for nonoliguric AKI. Common etiologies were eliminated following an adequate clinical evaluation, and renal ultrasound showing no abnormality. The fractional excretion of sodium (FENa) was 2.64% without urine dipstick anomalies. Based on these findings, no urine microscopy was performed at that time. The patient had no exposure to non-steroidal anti-inflammatory drug (NSAID) or intravenous contrast. Relevant laboratory measurements during the first 5 days of hospitalization are shown in Figure 1. The first result of serum uric acid, taken 36-hour after the convulsive episode, demonstrated hyperuricemia at 782 µmol/L (reference range: 206-441 µmol/L) and a slight CK elevation, with no acidosis on blood gas at that time. The uric acid to creatinine ratio on urine was 0.57 (reference range for urate nephropathy: ≥1.0 mg/mg (13)), but was collected only at day 3 while the patient was already receiving intravenous hydration. Since the severe hyperuricemia could not be explained by a prerenal disease with increased tubular reabsorption 14 and no other AKI etiology seemed compatible, a presumptive diagnosis of uric acid nephropathy was made.

Timeline of laboratory results and treatments during hospitalization of patient #13 (index case).
Urine alkalinization treatment based on intravenous bicarbonate (150 mmol/L) was quickly initiated until the value of uric acid fell in the reference range. Target urinary pH was maintained ≥7.0 during treatment with urine output of 5 to 6 L/day during the first 3 days. Allopurinol was initiated at the same time. Uric acid level and kidney function rapidly improved. Noteworthily, a delayed increase in CK levels occurred 2 days after as showed in Figure 1, while creatinine was already going down. Intravenous hydration was changed to 0.9% NaCl, and the flow was adjusted to obtain a urine output of ~4 L/day to prevent additional CK-induced tubulotoxicity. Due to the rapid improvement of AKI once the treatment introduced, no renal biopsy was performed to confirm the suspected etiology. The patient was discharged at day-6 with a follow up at the outpatient nephrology clinic 3 months later where the SCr was 87 µmol/L.
Prospective Study
Methods and Design
In this observational study, all participants were recruited at the Center Hospitalier Universitaire de Montréal (CHUM) from August 2018 to September 2019, after being identified by neurologists or nephrologists. Participants were included if a GTCS occurred within 24 hours of study enrollment, defined as more than 5 minutes, or a series of seizures with incomplete return to baseline neurological status, as proposed by the International League Against Epilepsy. 15 Exclusion criteria were patients where consent was impossible and in whom a delay between blood sampling and last seizure was more than 36 hours. All included participants signed letters of informed consent. Ethics boards at the CHUM Research Center approved the protocol for this study. In addition to medical history, seizure characteristic, and demographics, urine and blood samples were prospectively collected daily from Day 0 to Day 3 for measurement of lactate, blood gas, CK, SCr, serum uric acid, and urate-to-creatinine ratio. All analyses were performed at the CHUM hospital central laboratory. Day 0 samples were collected as soon as the consent was obtained. The sample size initially planned was 30 patients, but because recruitment was slow and challenging, the study was stopped prematurely after 1 year. Only descriptive statistics were used (SPSS 25 Software, IBM Corp, Armonk, NY).
Results
A total of 17 patients were approached to participate in the study after being initially identified following a severe seizure, 12 patients met inclusion criteria for further analysis as shown in Figure 2. Baseline characteristics are depicted in Table 1. One patient deviated from protocol inclusion criteria but was included because his seizure, initially categorized as focal, was described as a severe tonic-clonic left upper and lower body convulsion of more than 20 minutes without loss of consciousness. The investigators therefore considered this seizure event severe enough to be included. The median seizure duration was 5.0 minutes (interquartile range [IQR], 2.0-12.5). All patients with seizure duration of less than 5 minutes have had a series of seizure with an incomplete return to baseline neurological state. Two patients were on maintenance dialysis; their SCr values were not included into the analysis because variations were most likely due to dialysis schedule rather than postseizure kinetic.
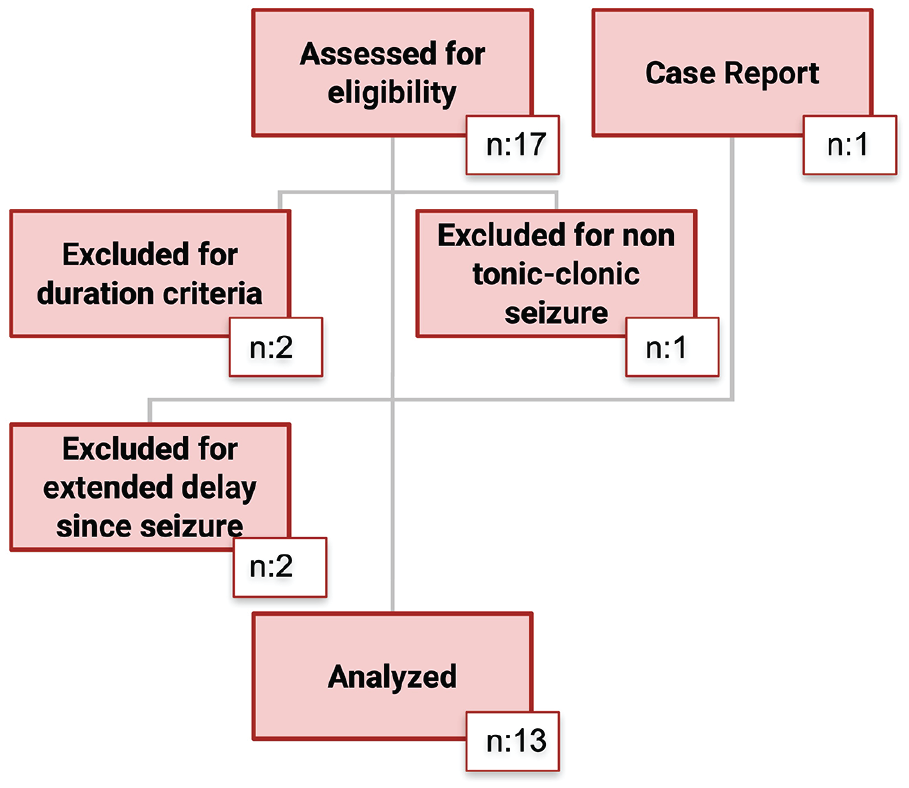
Flowchart and overview of included patients.
Baseline Characteristics of All Patients.

Interquartile range.
Using Chronic Kidney Disease Epidemiology Collaboration Equation (CKD-EPI). Excluding two (2) patients on maintenance dialysis (n = 11).
Excluding the patient presented in the case report (n = 12).
Uric acid and SCr were measured at least two times in the first 48 hours for all patients (Figure 3). No patient received uric acid lowering drug, excluding the index case. As shown in Table 2, from day 0 to day 3, the median uric acid level decreased from 346.0 μmol/L (IQR, 155.0-377.5) to 178.0 μmol/L (IQR 140.0-297.5). However, maximum uric acid levels remained within normal ranges in all but two (15.4%). In patients other than our index case (#13), increases in SCr were relatively mild or absent. However, the median SCr also was higher at day 0 (73.0 µmol/L [IQR, 51.0-80.0]) compared to day 3 (57.0 µmol/L [IQR, 44.0-70.0]). Considering the lowest creatinine value as baseline, four patients (36.4%) classified as having at least stage-1 AKI based on elevation in SCr more than 1.5 times the baseline as per Kidney Disease: Improving Global Outcomes (KDIGO) criteria. 16 Measures of urine urate-to-creatinine ratio were available for all patients within 36 hours following the last reported seizure. No patient had a urinary urate-to-creatinine ratio ≥1.0, which has been proposed as an indicator of risk of urate nephropathy, 13 and no notable variation was observed through the follow-up duration. Six patients (46.2%) had CK levels higher than the reference value for their gender. The median maximum value of CK was 218.0 units/L (IQR, 118.5-468.0) and remained within reference range, without signal toward a particular kinetics; 6 patients (46.1%) had their maximal value at day 1 and the remaining at day 2 or 3. Four patients had lactate ≥2.4 mmol/L, and two patients (#7 and #13) had profound metabolic acidosis related to severe status epilepticus.

Laboratory values according to the time after seizure: Line graphs represent serum uric acid (A) and serum creatinine (B) for each consecutive participant number (#).
Laboratory Measurements During Follow-Up for All Patients.

Note. IQR = interquartile range; KDIGO = Kidney Disease: Improving Global Outcomes; CK = creatine kinase; AKI = acute kidney injury.
Day 0 represents the first measurement available after the index seizure.
Negative value indicates decrease during follow up.
All data after exclusion of patients on maintenance dialysis (n = 11).
According to AKI KDIGO criteria, using the minimal observed creatinine during observation as the baseline value.
After unit conversion into mg/mg.
Discussion
Acute hyperuricemia is caused by an increase of purine liberation, which can be the result of increased cellular turnover, cell lysis following aggressive cancer chemotherapy regimens or even severe rhabdomyolysis. 10 Humans are particularly affected because, through the ape evolution, we lost the uricase enzyme allowing its conversion into soluble allantoin—the result is that uric acid has become the end-product of purine degradation. 17 If the amount of uric acid in the urine exceeds solubility threshold, the deposition and accumulation of uric acid crystals in renal tubules might lead to acute urate nephropathy.10,13 A combination of factors altering the balance between production and elimination of uric acid could explain seizure-induced acute hyperuricemia. After an excessive release of nucleosides by hypoxic tissues that are transformed into uric acid by xanthine oxidase,10,18 the metabolic acidosis from lactate generation and respiratory hypoventilation creates acidic urine making uric acid less soluble, increasing its precipitation into tubular lumen. 10 In addition, hyperthermia and profuse sweating from the adrenergic state during a seizure might favor dehydration and increase tubular water reabsorption, increasing tubular urate concentration. 9 Other factors as relative renal ischemia due to shunting of blood flow from visceral organs to muscles during convulsion may also contribute to kidney injury and uric acid accumulation. Jiang et al previously demonstrated, in an ER setting retrospective study, that uric acid levels were significantly higher in 179 patients with convulsions compared to 212 nonepileptic neurological emergencies (p < .001; 7). However, no correlation with creatinine was made, and the severity of these seizures is unknown. Recently, Nass et al published the first prospective study that investigated metabolic markers in a cohort of patients who had had provoked tonic-clonic seizures in an EEG monitoring setting. They showed that serum uric acid increased by 61.1% (p < .001) 2h after convulsion and usually returned to baseline levels after >24 hours. However, in this cohort, only six of the 32 patients had had a true GTCS. Likewise, all the patients were recruited in the context of elective EEG monitoring, making this young population difficult to generalize to patients truly at risk of developing AKI.
Our index case highlights the risk of severe hyperuricemia and AKI following GTCS. In the absence of other cause of AKI, based on elevated uric acid level checked even 36 hours after presentation and normal serum CK, it seems likely that AKI was related to urate nephropathy. Unfortunately, uric acid was not measured within the 24 hours after GTCS, allowing the possibility that the actual peak value was missed and no urine microscopy was performed to confirm diagnosis. Interestingly, this patient had a previous episode of unexplained AKI following GTCS; even though a similar episode of urate nephropathy could have occurred at that time, this remains a possibility, which we cannot confirm because no uric acid level was measured. It remains unknown why some patients may be more prone to develop severe hyperuricemia and urate nephropathy in context of seizure, or if the index case had additional metabolic abnormalities making him more at risk for developing such complication.
Our prospective case series, conducted in a tertiary teaching hospital, investigated SCr and uric acid kinetics in a pragmatic context of severe seizure. It is the first study that included a follow up of more than 48 hours, and a population from difference sources, not only patients electively admitted for EEG monitoring. However, a posteriori, recruiting patients who had unprovoked convulsive episodes at home—which caused notable delay between the convulsion and the first blood sample—probably did not allow us to capture the uric acid peak level, as demonstrated later by Nass et al. Despite this limitation, our results are compatible with conclusions reported in previous studies.3,7 We found higher uric acid levels at the first sample compared to subsequent values during follow-up. However, the magnitude of increase in uric acid levels in our cohort was mild (22.9% median increase) and lower than the increase reported by Nass et al in the setting of EEG monitoring induced-seizure (61.1%, p < .001), where the kinetics of uric acid showed a peak value 2 hours after the seizure episode. The limited increase in serum uric acid noted in our study could be explained by a median sampling delay of 10 hours. The same observation might apply to the urine urate-to-creatinine ratio which correlated with the serum value at the sampling time.
As a significant proportion of uric acid excretion is renal, baseline kidney function is a major factor that can affect the uric acid level, the ability to remove additional urate load as well as the risk of AKI. We decided to include two patients on maintenance dialysis in the acid uric analysis because they were still at risk of developing acute hyperuricemia following GTCS. However, when excluding these two patients from the data set, the uric acid level at day 0, 346.0 μmol/L (IQR, 172.0-390.0) and day 3, 178.0 μmol/L (IQR, 140.0-297.5) remained similar to results previously reported for all patients.
Release of CK and lactate following severe seizures is well described in the literature, and it is historically taught that it correlates with the episode severity. 1 However, in our cohort, in exception to our initial case reported (#13), as well as in other cohorts2,3,7 CK elevations were mild and the median maximum value of CK remained within reference range. Solely, these elevations were unlikely to increase the risk of AKI. Nephrotoxic CK elevation is not frequent following GTCS, which limits its physiological explanation of AKI and its sensitivity as a diagnostic maker of seizures in ER. 2
Another mechanism may drive the link between uric acid and AKI in the context of seizure. Acute hyperlactatemic metabolic acidosis following GTCS, by a rapid decrease in urine pH and tubular flow, could lead to a significant reduction of urine solubility threshold. In this context, even moderate increases in uric acid levels may result in an acute supersaturation and formation of urate crystals into the tubular lumen.10,13 Since no urine microscopy was carried out during the study due to limited resources and no prospective biobanking, we could not verify this phenomenon. The urine urate-to-creatinine ratio were all negative and did not change over time, including for the initial case reported. Interestingly, some authors suggested that the urate nephropathy diagnostic ratio cutoff historically recommended should be reduced, particularly in young patients with normal proximal tubular function.19,20
We showed, despite a limited sample size, that four patients (36.4%) had an increase in creatinine compatible with at least stage-1 AKI. Interestingly, in reviewing the medical charts of these patients, no summary of hospitalization mentioned the occurrence of AKI excluding for patient #13. Our index case apart, the magnitude of the creatinine increase was mild, and the clinical significance of these findings remains unclear. Therefore, our results do not support a correlation between these creatinine elevations and hyperuricemia. Similarly, not performing urine microscopy might be a limitation. Several confounding factors frequently associated with AKI could have been be involved, such a medication, baseline kidney function, and medical comorbidities. The design and the sample size of our study did not allow us to control for that. Larger studies are needed to confirm this association.
Like tumor lysis syndrome, where risk factors are well known and where monitoring recommendations and preventive measures have proven their effectiveness 21 ; seizure-induced hyperuricemia and its associated management should be further investigated.
Conclusion
This report highlights the phenomenon of hyperuricemia following severe seizures. Although the number of patients recruited was limited, our study is the first to pragmatically describe the kinetics of uric acid and creatinine following severe seizures in patients recruited in various settings. A signal toward an acute increase of SCr and uric acid levels was observed, before returning to baseline. However, the observational design, the limited sample size, the absence of urine microscopy and the delay before blood sampling do not allow to confirm causality. Urate tubular deposition is a preventable form of nephrotoxicity where treatments are already accessible, such as urine dilution with intravenous fluids, urine alkalinization, and hypouricemic therapy. Quickly identifying these at-risk patients remains challenging. Until we have more evidence regarding the risk of urate nephropathy following severe seizures, we suggest that uric acid measurement could be included in the initial management of severe seizure. Further larger studies are required to confirm the effectiveness of such interventions in AKI prevention.
Footnotes
Acknowledgements
The authors would like to thank the patients who consented to participate and the CHUM Nephrology service, which offered all of the funding necessary to carry out this study and publish it. The authors would also like to thank the neurology department and Marie-Line Caron for their precious help for recruitment.
Ethics Approval and Consent to Participate
Formal Research Ethics approval was obtained. Consent was obtained for all participants.
Consent for Publication
All authors consent to the publication of this study
Availability of Data and Materials
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: All financial support for this research and publication of this article came from the CHUM Nephrology department. J.M.C. elaborated this article while receiving scholarship funding from the Fonds de Recherche en Santé du Québec, Société Québécoise de Néphrologie and the Académie CHUM.