
Abstract
Purpose of review:
The current review will discuss on the progress of studying the transition phase between acute kidney injury (AKI) and chronic kidney disease (CKD) through improved animal models, common AKI and CKD pathways, and how human studies may inform different translational approaches.
Sources of information:
PubMed and Google Scholar.
Methods:
A narrative review was performed using the main terms “acute kidney injury,” “chronic kidney disease,” “end-stage renal disease,” “animal models,” “review,” “decision-making,” and “translational research.”
Key findings:
The last decade has shown much progress in the study of AKI, including evidence of a pathophysiological link between AKI and CKD. We are now in a phase of redesigning animal models and discovering mechanisms that can replicate the pathological conditions of the AKI-to-CKD continuum. Translating these findings into the clinic is a barrier that must be overcome. To this end, current efforts include prediction of AKI onset and maladaptive repair, detecting patients susceptible to the progression of chronic maladaptive repair, and understanding shared signaling mechanisms between AKI and CKD.
Limitations:
This is a narrative review of the literature that is partially influenced by the knowledge, perspectives, and experiences of the authors and their research background.
Implications:
Overall, this new knowledge from the AKI-to-CKD continuum will help bridge the discontinuity that exists between animal models and patients, resulting in more effective translational biomarkers and therapeutics to test in known AKI pathologies thereby preventing the chronicity of kidney injury progression.
What was known before? (why is this review important)
Acute kidney injury (AKI) is a common complication of acute illness and a growing public health concern. Much progress has been made in understanding the pathogenesis, progression, and repair of AKI, but unfortunately few therapeutic agents have improved patient outcomes.
What this adds
Review of potential strategies to translate knowledge of AKI into the prevention of chronic kidney disease and end-stage renal disease at the bedside, with an emphasis on both traditional translational research (bench to bedside) and reverse translational research (starting with human studies to inform mechanisms that are validated in the laboratory setting).
What are the key messages?
More knowledge of human AKI at the tissue level is needed along with validation of these discoveries in multiple animal models that mirror the complexities of human AKI. Promising therapeutic agents should then be tested in high-risk patients with known causes of AKI in whom the timing of AKI onset or transition to maladaptive repair can also be determined. Teamwork and stakeholder engagement will be critical to these efforts, which must involve patients given the increasing emphasis on human tissue.
Introduction
Acute kidney injury (AKI) is a common complication of acute illness that affects as many as 20% of hospitalized patients.1,2 While the in-hospital morbidity and mortality associated with AKI is well-described, 3 it is now clear that individuals who survive to leave the hospital after an episode of AKI are at persistent risk of adverse kidney outcomes.4-6 For example, a meta-analysis demonstrated that compared to patients without AKI, survivors of AKI have an almost 10-fold greater risk of developing de novo chronic kidney disease (CKD) and a 3-fold greater risk of end-stage renal disease (ESRD). 6 Based upon an AKI population incidence of 2147 per million people, it is estimated that in the developed world (United States, Canada, Europe, and Australia/New Zealand), there will be more than 2 million cases of AKI annually, of which 3% to 15% will develop ESRD and 20% to 50% will progress to CKD. 7 The majority of these cases will be due to decreased kidney perfusion (i.e., pre-renal, sepsis, cardiac failure) and nephrotoxic medications, with small overall contributions from glomerulonephritis, interstitial nephritis, and obstruction.8,9 The incidence of AKI has increased over the last 15 years and is expected to double over the next decade, 7 suggesting that AKI and its association with progressive CKD and ESRD represents a growing public health burden.
Clearly, the lack of effective therapeutic interventions for the prevention or treatment of AKI is a major unmet medical need. Several workshops have recently assembled to identify and overcome barriers in the translation of basic knowledge of AKI into the prevention of CKD and ESRD.10-13 Much progress has been made in understanding the pathogenesis, progression, and repair of AKI, which has resulted in numerous potential therapeutic agents to evaluate. However, AKI has many different etiologies, and the timing of its onset in humans is often unpredictable; these latter factors have minimized the human health impact of basic knowledge gained from preclinical and animal models of AKI. Notwithstanding these challenges, substantial optimism exists that past “failures” will soon produce effective interventions to prevent and treat AKI thereby lessening its long-term sequelae. Although there are several approaches to describe the progression of AKI research, this review will be limited to detail some key reasons for this positive outlook, focusing on the current state and barriers affecting AKI research, recent examples of successful therapeutic interventions, and how these lessons may inform future AKI translational research.
Current State of Basic Science Research
One of the major difficulties we face in understanding the kidney is narrowing the knowledge gap that exists between animal model discoveries and clinical observations. Partly, AKI is difficult to reproduce within animal models given its complexity and multifaceted etiology. Over the years, animal models have improved from defining a specific injury with a unique maladaptive repair response of fibrosis to recent animal studies proposing that the repetitive exposure and variation in dosages are fundamental in influencing the outcomes of AKI and its continuum into CKD, regardless of the initial insult.11,14 These AKI-to-CKD transition models are capable of recapitulating the pathological mechanism of maladaptive repair that commences in the tubular region during an AKI response, reduces kidney structure and function, and propagates into interstitial fibrosis throughout CKD. This is translatable to human studies because the severity of injury can influence a variety of integral repair mechanisms to be continually activated in a maladaptive fashion from AKI to a CKD response. For example, continual expansion of activated fibroblasts, which are the effector cells of fibrosis, is found within AKI and CKD due to a persistent injury signal irrespective of the type of injury. 15 This is also the case with reduced capillary density, which is found in both AKI and CKD and leads to hypoxia-induced fibrosis. 16 Full comprehension of these common pathological mechanisms between AKI and CKD can fashion an approach to better target the progression of CKD in the post-AKI setting.
Conventional animal models used to study the AKI-to-CKD transition have been designed through modification of the ischemia/reperfusion injury (IRI) and nephrotoxicity models. In addition to the dosage of injury, these redesigned animal models are also established on the timing of injury so as to develop effective therapies that are based on a rationale for targeted treatments at specific time points between the AKI-CKD continuum. Consequently, such models will help determine dosage-responsive targets and crucial time points needed to develop effective therapies of intervention. Overall, the following described IRI and nephrotoxicity models are designed to permit the kidney to recover from an initial insult by triggering de-differentiation and proliferation of the remaining damaged cells following an episode of AKI, but enter CKD with a more severe injury based on longer timing and higher/repeated dosage of injury.
IRI Models
One of the main causes of AKI is IRI due to various clinical procedures of clamping of vessels during renal transplantation and postoperative decrease in perfusion. Multiple species have been used to recapitulate IRI for specific requirements. Larger species (pigs and dogs) are used for their resemblance in anatomy and size to humans for surgical-like procedures that induce IRI. Rodents are commonly used to study IRI because of the availability of genetically modified models and their ability to generate large sample sizes due to short breeding periods. IRI is a simple and reproducible procedure induced by temporarily clamping the arteriole blood flow and restoring it for redistribution of oxygen and nutrient delivery.17,18 Within mouse models, the kidney hilum is clamped between 30 and 60 minutes followed by reperfusion for days to weeks to cause a post-ischemic response reflective of an AKI to CKD transition.17-19 This impairment subsequently leads to the acute pathological changes of early inflammation, tubular dilatation and necrosis, significantly elevated creatinine levels and brush border loss within days to chronic events of sustained inflammation, interstitial fibrosis, and tubular atrophy.17,20 Body temperature fluctuation is one of the most important factors affecting the severity of experimental IRI, that is, lower temperatures cause less damage; therefore, a homeothermic monitor system and heating pad must be used to accurately keep mice warm during surgery.18,21,22
Nephrotoxic Models
By virtue of its primary role in clearing toxicants from the blood, the kidney is inherently vulnerable to nephrotoxic injury due to its continuous exposure to such harmful contaminants. Several nephrotoxic models have been developed to generate injuries throughout the AKI-to-CKD transition toward CKD progression using various repeated treatments and/or dosages of toxins, such as folic acid (FA), cisplatin, and aristolochic acid (AA). FA is a simple injury model commonly performed on rodents using a dosage and timing with toxicokinetics comparable to those associated with disease in humans.23,24 FA is a nephrotoxicant which crystalizes in the epithelial tubules at high doses. Consequently, this results in tubular lesions, intratubular crystal-obstruction, and a direct toxic effect on the epithelial cells, 25 accompanied by mild fibrosis in the chronic phase. Remarkably, these same injury characteristics are found in patients with AKI, suggesting the FA-injury model to be relevant to human AKI mechanisms. 26 Cisplatin is a widely used chemotherapy drug with a nephrotoxic effect that is cumulative and dose-dependent. 27 The notable pathophysiological effects of cisplatin include proximal tubular and vascular injury and necrosis leading to chronic inflammation and fibrosis reflective of CKD development.28-32 AA is a chemical agent commonly found in the Aristolochiaceae plant family that has nephrotoxic effects. Notably, the AA model offers a more directly translatable opportunity to study nephrotoxological mechanisms in humans, as environmental exposure to AA represents a significant threat to human health. 33 AA nephrotoxicity was initially reported in a Belgian patient cohort who ingested slimming pills containing this plant extract. 34 Despite several Food and Drug Administration (FDA) warnings, numerous ingredients contain AA in traditional medicine from China, Japan, and India. 34 Furthermore, there is a high incidence of nephropathy in the Balkan region suspected to be due to AA contamination that occurs during their wheat grain harvest. 34 Although the exact mechanisms of AA-dependent nephrotoxicity are not fully understood, G2/M arrest is considered to be a major pathological mechanism of cortical tubular atrophy and extensive interstitial fibrosis.34-36 In these different types of toxin studies, each have their respective controlled dosage and timing of toxin-induced injuries which enables researchers to investigate mechanisms that are associated with features specific to the AKI-to-CKD transition.
Numerous signaling pathways of kidney repair are shared between AKI and CKD, whose continuous activation contributes to their interconnected pathogenesis. Those mechanisms underlying severe or episodic AKI injury are continually activated to progress into and contribute to the state of CKD, while these similarly activated pathways in CKD may increase the sensitivity of the kidney to successive AKI insults.15,16,37 Well-known signaling pathways activated during AKI that are chronically activated into CKD include senescence, inflammation, epigenetic modifications, and developmental pathways.
Cellular Senescence and Associated Pathways
One of the earliest response pathways that is activated in injured tubular epithelial cells (TECs) from an AKI model is p53-mediated G2/M cell cycle arrest, which consequently leads to the program of senescence.20,38,39 Cellular senescence is involved in normal wound healing by producing a distinct secretome, termed the senescence-associated secretory phenotype (SASP), of pro-repair and pro-inflammatory factors. 40 In particular, G2/M-arrested tubular cells were noted for their increase in c-jun NH2-terminal kinase (JNK) signaling, which is known to enhance the gene transcription of the major profibrotic factor, TGF-β1.38,41 Although the expression of TGFβ is needed to stimulate proliferation of tubular cells for regeneration during early injury states of AKI, the prolonged state of cell cycle arrest in CKD results in a TGFβ signaling pathway that is continuously activated to induce the chronic renal fibrotic events of fibroblast activation and extracellular matrix production.40,42 Furthermore, prolonged TGFβ can also contribute to the senescent phenotype of renal proximal TECs by stimulating IGF-binding protein 7 (IGFBP7), 43 which is a component of the SASP. 43
Inflammation Pathways
Pro-inflammatory factors are important constituents of the SASP during the communication of the senescent tubule to its neighboring cells, in particular by producing interleukin 6 (IL-6) and TNF-α. Within the AKI model, the IL-6 signaling axis was implicated in activating an injurious inflammatory response, 44 whose chronic activation led to CKD fibrotic features. 45 In the cisplatin AKI model, TNF-α was shown to play a central role in the activation of pro-inflammatory cytokine response, 46 while two separate studies confirmed the association of circulating TNF-α and its receptors with progression of CKD.47,48 In the nephrotoxic and ischemia models of AKI-to-CKD transition, the chronic pro-inflammatory state involved not only elevated IL-6 and TNF-α, but also the inflammatory markers HMGB1, CSF-1, and MCP-1. 43 In addition to senescence, programmed death of TECs is a hallmark of AKI known to regulate inflammation and promote repair. Although TECs undergo apoptosis and necrosis, the predominant type of tubular death during AKI is necroptosis.49-51 Dead-cell debris is involved in triggering an inflammatory response but its efficient removal is necessary to restore tubular structure and function after AKI. In fact, the apoptosis inhibitor of macrophage (AIM) was shown to be critically involved in the kidney injury molecule-1 (KIM-1)–associated clearance of cell debris, facilitating the resolution of IRI-induced AKI. 52
Epigenetic Pathways
Diverse epigenetic mechanisms are known to promote pro-inflammatory and profibrotic gene expression. The most common epigenetic modifications seen with acute and chronic kidney diseases include DNA methylation and histone acetylation/deacetylation.53-55 An IRI AKI-to-CKD transition mouse model provided evidence of the overlapping mechanism of progressive histone modifications at specific pro-inflammatory/profibrotic genes, such as MCP-1, TGFβ, and collagen. 56 A common epigenetic modification during AKI involves a TGFβ-driven hypermethylation and silencing of the RASAL1 gene, an inhibitor of RAS. Consequently, RAS is continuously activated to drive fibroblast activation and proliferation, which overtime induces an CKD transition. 57 Furthermore, inhibitors of histone deacetylation were shown to attenuate kidney injury and the development of AKI-to-CKD in models of AA-, cisplatin-, and ischaemic-induced kidney injuries, potentially describing the importance of regulating epigenetic outcomes during the transition of AKI to CKD.58-61
Developmental Pathways
Injured tissues enter a regenerative phase that recapitulates key developmental pathways. 62 Wnt and Notch signaling are canonical pathways that regulate morphogenesis and progenitor populations during development. Expression of Wnt and Notch diminishes once nephrogenesis is complete; however, during an injury, the kidney epithelium reactivates the Wnt and Notch pathways to re-establish the tubular microanatomy. Both Wnt and Notch are upregulated during AKI; however, their sustained activation drives AKI-to-CKD progression by promoting fibroblast activation and fibrotic matrix deposition.17,63,64 Specifically, Wnt4 ligand signaling through β-catenin stabilization is sufficient to drive the myofibroblast marker αSMA in kidney interstitial stromal cells in the absence of an injury signal, resulting in these effector cells to drive fibrosis in uninjured kidneys. 65 In the case with Notch signaling, the Notch-2 receptor and its ligand Delta-1 have been implicated in the proliferation of renal tubules during AKI, 66 while Notch-1 and Delta-4 play key roles in fibrosis development.67,68
In summary, advancing our animal models to reflect the clinical progression of AKI to CKD is needed to delve into the mechanisms of action and pave the way for improved clinical translation. As we continue to develop and refine our AKI-to-CKD models and identify their continuum pathways, we will be in a better position to clarify their causal relationship for preclinical testing of potential diagnostic and therapeutic approaches.
Transition From Animal Models to Patients
Animal models provide the means to accelerate the discovery and evaluation of targets and pathways, which are then assessed in patients on whether they are modulated and serve as primary drivers of pathogenesis. As discussed above, animal models of the AKI-to-CKD transition have provided a better understanding that the pathways of senescence, inflammation, epigenetics, and development are implicated in the continuum between both states of kidney disease. As such, targeting the mechanisms of these pathways has potential for intercepting the transition from AKI to CKD. Several promising targets have been discovered from such AKI-to-CKD models. For example, p53 inhibitors have shown promise of inhibiting senescence and SASP in preventing the progression from AKI to CKD.20,69 Apart from acting as potential therapeutic targets, molecules from these pathways could also serve as biomarkers since current methods are imperfect and poorly reflect the measurement of kidney function. For example, some traditional methods that evaluate the degree of nephropathology, which include measuring GFR by creatinine levels and fibrosis by collagen deposition, detect kidney injury at stages when there has been sufficient damage. However, translation of AKI-to-CKD relevant biochemical pathways offers potential for effective therapies. Specifically, detection of subtle changes in genetic modifications and molecular pathways implicated in the AKI-to-CKD continuum during the early stages of AKI before their transition into CKD could offer better preventative measures against the irreversible states of kidney damage. For example, studies involving patients with AKI have been reported to have genes with DNA methylations and histone modifications that altered their transcription and subsequently implicated in renal injury.70,71 Therefore, future studies are needed to delineate the timing of AKI-to-CKD transition pathways to optimize any of their protective effects but prevent the outcomes of their chronic activation. Moreover, genomic and proteomic techniques would offer an opportunity to discover, validate, and gather the dynamic expression of numerous targets and better grasp their role in the pathogenesis of kidney injury.
Overcoming Limitations
Progress has been made in developing mouse models that closely relate to the progression of CKD from AKI in patients. There is a need to find better strategies to identify therapeutic targets from studies coming from animal models that can relate to human nephropathologies. This is a major concern because of the high cost of conducting clinical trials. One way to increase the chances of a translatable target into humans could potentially come from targets that have conserved effects and/or pharmokinetic properties across various species. Furthermore, the study of multiple AKI-to-CKD transition models may help identify conserved pathways in the AKI-to-CKD continuum. Animal models also tend to have limitations in reproducing, or at the very least resembling, human disease due to differences in physiology/pathophysiology between animals and humans, and also to faulty experimental procedures or models that simplify insults for complex clinical states. A solution would be to use emerging concepts from patient data as a guide to recapitulate expected results by fine-tuning animal models. As a result, the in vivo modeling of patient data would permit a lower occurrence of false positives and higher certainty of therapeutic targets emanating from detailed mechanisms. Overall, studies should be rigorously designed and executed, taking into consideration strain background, diet, and other relevant variables, and be conducted in a blinded manner.
Current State of AKI Clinical Research
To take full advantage of the animal models and mechanisms described above, subsequent studies in humans should closely resemble the conditions in which the therapeutic interventions were initially discovered. This creates challenges because the onset of AKI and transition to CKD in humans can be difficult to recognize, and the mechanism used to induce injury in animal models may not replicate the multiple etiologies responsible for AKI and CKD progression in humans. To this end, much progress has been made in the ability to identify and predict patients at high risk for AKI and CKD progression. Koyner et al 72 integrated a multicenter risk algorithm into the electronic health record of 202,961 hospitalized patients over a 4-year period. Using routinely collected data readily available in the electronic chart, patients at risk for Kidney Disease Improving Global Outcomes (KDIGO) stages 1 and 3 AKI were identified in real-time, a median 42 and 35 hours before AKI onset. For patients at risk for CKD progression, James et al 73 developed a six-variable prediction equation in 9973 patients. Readily available online and remotely (qxmd.com), this simple tool accurately identifies progression to stage 4 CKD within 1 year of hospital discharge. Both studies highlight that there is sufficient time to deliver therapeutic interventions to patients at high risk for AKI and CKD before these events occur.
Progress has similarly been achieved in the rapid identification of patients with established AKI, though less so with CKD progression after AKI. Currently, the detection of AKI relies primarily on serum creatinine concentration and urine output 74 ; however, there are few symptoms that accompany AKI and CKD which often leads to delays in the recognition and treatment of these conditions. The introduction of electronic creatinine alert systems for AKI, which notify healthcare providers within minutes of creatinine elevation, helps address this problem. These alert systems have been scaled nationally in the United Kingdom, demonstrating their feasibility. In a multicenter study of 24,059 AKI episodes, an electronic creatinine alert coupled with a care bundle increased the detection of AKI, medication reviews, and fluid assessments. 75 While this UK study and another study in the United States of 2400 patients failed to reduce AKI severity and mortality,75,76 they demonstrate the capacity to rapidly identify established AKI in real-time. The disappointing results may relate to the insensitive nature of serum creatinine; it is non-specific and only elevates after injury has already occurred, which is a principle that also applies to the identification of maladaptive repair after AKI.
Accordingly, biomarkers have been sought to aid in detection of subclinical injury, provide information on the etiology and anatomic location of AKI, detail the extent of damage, and measure ongoing injury and/or repair/fibrosis after AKI. 77 Biomarkers of tubular injury such as IL-18, 78 neutrophil gelatinase-associated lipocalin (NGAL), 79 and KIM-1 80 can differentiate between pre-renal azotemia and acute tubular injury, but their modest discriminative ability has limited their translation beyond clinical trial recruitment.81,82 The FDA recently approved the first AKI point-of-care biomarker device called NephroCheck. 83 Its positive predictive value to diagnose KDIGO stage 2 or 3 AKI is 49% and the negative predictive value is 97%, suggesting its strength might lie in the ability to identify patients at low risk for severe AKI.83,84 NephroCheck measures urinary levels of tissue inhibitor of metalloproteinase-2 (TIMP-2) and IGFBP7, which are cell cycle arrest biomarkers. It is hypothesized that TIMP-2*IGFBP7 may also serve a protective effect on the kidney by minimizing the proliferation of damaged cells. As for CKD progression after AKI, there remains no FDA-qualified biomarker to measure continued injury or intrarenal fibrosis, which limits the detection of maladaptive repair and the potential efficacy of treatments that target these pathways. Other options for fibrosis measurement include magnetic resonance imaging (MRI) techniques, such as blood oxygen level–dependent MRI, 85 fibrosis-specific contrast agents, 86 and elastography techniques 86 whose clinical utility remains to be fully determined.
The above progress in primarily the recognition and diagnosis of AKI can help identify the challenges and opportunities that remain to translate basic knowledge of AKI into the prevention of CKD and ESRD at the bedside. First, most risk scores do not address the etiology of AKI or AKI subtypes/endophenotypes; the latter terms being the combination of clinical features, comorbidities, genetics, biomarker expression, and population-specific factors that contribute to its onset and progression. 10 Even if high-risk patients for AKI and its sequelae can be identified early, therapeutic interventions are likely to be dependent on the cause or subtype of AKI. To address this obstacle, one suggested strategy is to focus on animal and human populations in whom the etiology of AKI is established, such as cardiac surgery, coronary angiography, and chemotherapy. This work will require kidney tissue from human samples with well-characterized causes and courses of AKI, which is currently underway by the National Institutes of Health (NIH).10,87 In addition to the right patient population, potential therapeutic interventions need to be administered at the right time. Delays may explain the lack of treatment benefit for electronic creatinine alert systems, but they also affect translational studies, such as in a multicenter study of recombinant IGF-1 that was administered as late as 6 days after the onset of AKI. 88 This barrier reinforces the importance of concentrating research to causes of AKI with precise onsets, which is challenging for CKD progression after AKI where the exact timing of maladaptive repair remains poorly defined. It is possible that this gap may be filled by biomarkers to identify the transition to repair and fibrosis after AKI. However, AKI biomarkers have yet to realize their full potential. Their biologic actions are often not linked to therapeutic interventions, and their efficacy has traditionally been measured against serum creatinine rather than tissue samples or hard clinical outcomes. This practice has led the Acute Dialysis Quality Initiative to propose a framework that characterizes AKI using both functional and damage markers, 89 and the same principles could extend to CKD progression after AKI using the combination of creatinine, biomarkers, and MRI. It also remains unclear how biomarker expression patterns differ across subtypes of AKI. Through the Kidney Precision Medicine Project, the NIH is accumulating kidney biopsy samples in patients with AKI which may inform subtype-specific and common mechanisms to target. 87 It is unclear though how many of these samples will be from patients who develop CKD and ESRD after AKI. In the past, large interventional AKI trials have not followed patients long term, 90 and most nephrologists do not routinely follow patients after even dialysis-requiring AKI. 91 These latter points reinforce why much more is currently known about the prevention and treatment of hospitalized AKI, whereby the downstream consequences of CKD and ESRD may also be reduced.
A Path Forward?
Some of the aforementioned basic science and clinical barriers have been overcome, which may inform translational research strategies to improve outcomes for patients with AKI. Even in the absence of a targeted treatment, Meersch et al 92 reduced the absolute risk of AKI by 17% in patients undergoing cardiac surgery. This was accomplished by identifying high-risk patients with a validated cell cycle arrest biomarker (i.e., TIMP-2*IGFBP7) among a cohort with a single AKI etiology of known timing (i.e., cardiac surgery). The treatment in this randomized trial of 276 patients was a KDIGO bundle, comprising optimization of blood volume status and hemodynamics, avoidance of nephrotoxic drugs, and prevention of hyperglycemia. This study demonstrates how enrolling the right patient at the right time enhances the likelihood of finding an effect, as the number needed to treat would have been much higher than six had all 276 patients received the KDIGO bundle. 93 Not only does this study provide an example of AKI prevention that may be translated to other well-defined etiologies such as coronary angiography and chemotherapy, but it also demonstrates a successful strategy that could be replicated when testing potential treatments for established AKI and progression to CKD after AKI.
Continuing this progress will require stakeholder engagement from clinicians, basic scientists, trialists, industry, and the FDA, which is already underway. 10 One group conspicuously missing from these meetings is patients and their caregivers. Their involvement in AKI research could serve several functions, particularly idea prioritization, study design, and patient recruitment.94,95 Their role becomes even more important as expert groups recommend reverse translational approaches for AKI research to complement the traditional bench to bedside approach to treatment development. 10 In short, reverse translation involves working backward by first defining patient populations, clinical trial designs, and end points, as well as maximizing knowledge of human AKI with mechanistic studies requiring human tissue. Then, animal models are identified to replicate the above patient population and develop therapies. Reverse translation may require more commitment and risk from patients and their caregivers, and so before this work is undertaken it is critical to understand their perspectives on research strategies for AKI and CKD in addition to their willingness to support both traditional and reverse translational research.
Conclusion
The opportunity to make advances in the prevention and treatment of AKI and its sequelae has never been greater. There have been major advances in understanding the biology of AKI, CKD, and the clinical infrastructure to predict and diagnose AKI rapidly. Multiple workgroups have assembled,10-13 with different yet complementary translational strategies proposed to overcome the well-described challenges (Figure 1). Whether the focus is on individual AKI subtypes or common mechanistic pathways that drive CKD after AKI, more knowledge of human AKI at the tissue level is needed along with validation of these discoveries in multiple animal models that mirror the complexities of human AKI. Promising therapeutic agents should then be tested in high-risk patients with known causes of AKI in whom the timing of AKI onset or transition to maladaptive repair can also be determined. Biomarkers may help guide the timing of treatment delivery and should be incorporated in both the study of human and animal AKI. Teamwork and stakeholder engagement will be critical to these efforts, which must involve patients given the increasing emphasis on human tissue to guide AKI research. These new patient-centered research approaches and flexible translational strategies provide optimism that therapeutic interventions for the prevention and treatment of AKI will soon be available.
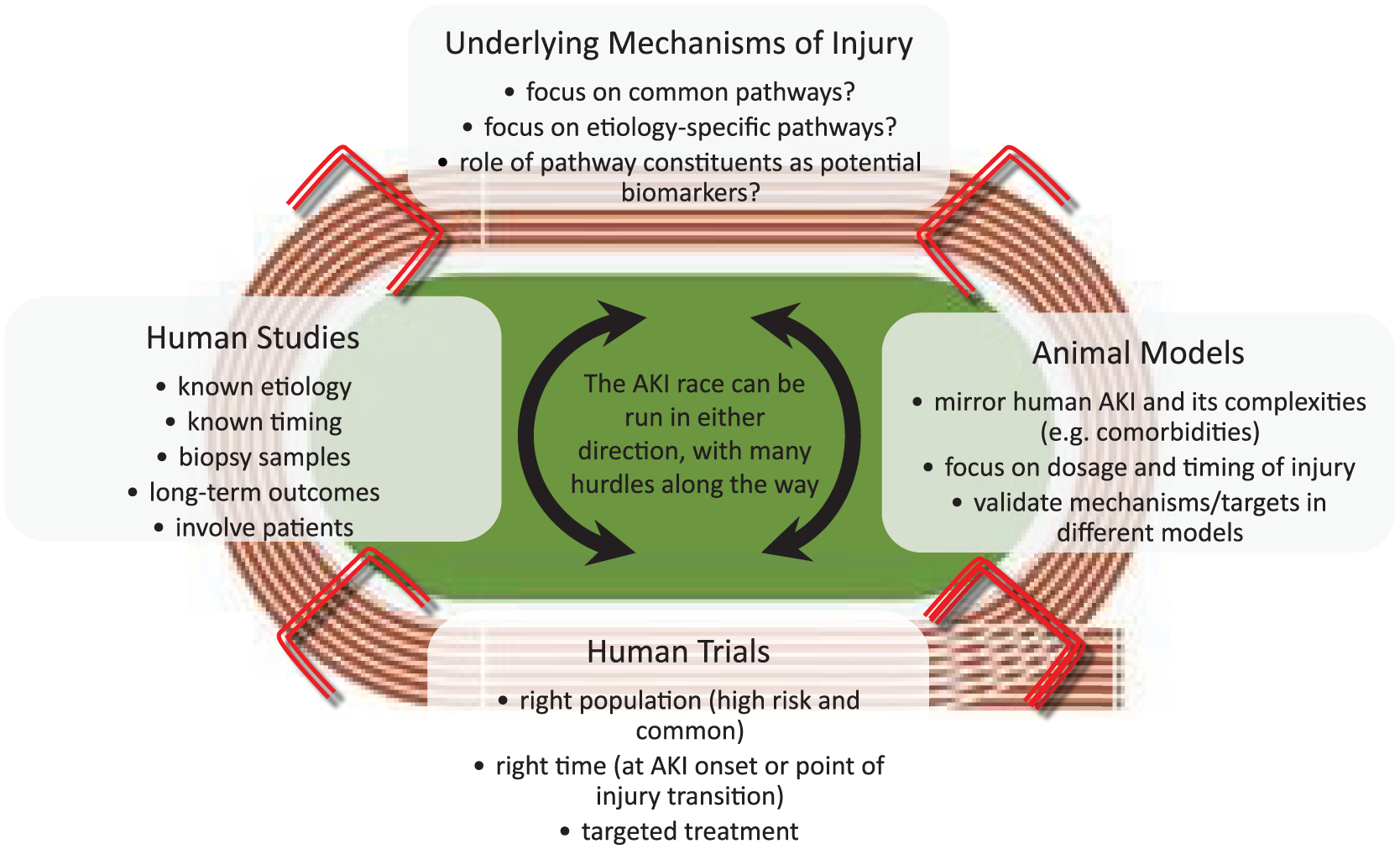
Potential strategies to translate knowledge of AKI into the prevention of CKD and ESRD.
Footnotes
Ethics Approval and Consent to Participate
This is a review article and does not involve any intervention on patients.
Consent for Publication
All authors consent to the publication of this research.
Availability of Data and Materials
This study is a review of previously published data and materials.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: S.A.S. and C.G. are supported by the Kidney Research Scientist Core Education and National Training (KRESCENT) Program New Investigator Award (co-funded by the Kidney Foundation of Canada, Canadian Society of Nephrology, and Canadian Institutes of Health Research).