
Abstract
Background:
Acute kidney injury (AKI) following cardiac surgery leads to increased morbidity and mortality. Characterization and validation of early biomarkers of AKI may ultimately facilitate early therapeutic intervention. We have previously identified that elevated urinary hepcidin-25 is inversely and independently associated with the development of AKI in adult cardiac surgery patients. Hepcidin-25 is an antimicrobial peptide that sequesters iron intracellularly, and its elevation following human ischemia reperfusion injury may represent a renoprotective response to minimize renal injury.
Objective:
Our goal was to validate urinary hepcidin-25 as a non-invasive biomarker in an independent cardiac surgery cohort, within the context of clinical AKI predictors.
Design:
Prospective observational cohort study.
Setting:
Adult cardiac surgery program at St. Boniface Hospital, Winnipeg, Manitoba, Canada.
Patients:
Adult cardiac surgery patients undergoing cardiopulmonary bypass (CPB), n = 306.
Measurements:
Urine hepcidin-25, measured on post-operative day (POD) 1.
Methods:
A prospective, observational cohort of adult CPB patients (n = 306) was collected with serial perioperative urine samples. Urine hepcidin-25 at POD 1 was measured by competitive ELISA. Its diagnostic performance was evaluated in conjunction with clinical parameters and the Thakar clinical prediction score, using multivariate logistic regression.
Results:
Urinary hepcidin-25 is elevated following cardiac surgery in AKI and non-AKI patients. Elevated urinary hepcidin-25 concentration was inversely associated with AKI on both univariate (odds ratio [OR]: 0.61, 95% confidence interval [CI]: 0.45-0.83, P = .002) and multivariate analysis (OR: 0.67, 95% CI: 0.50-0.95, P = .02). A combined model with clinical risk factors demonstrated that baseline estimated glomerular filtration rate (eGFR), diabetes mellitus, and urinary hepcidin-25 concentration had an overall area under the curve (AUC) of 0.82 (0.75-0.88) for predicting subsequent AKI development, which was superior to clinical prediction alone as determined by the Thakar score.
Limitations:
(1) A single-center observational study. (2) Polyclonal antibody–based competitive ELISA.
Conclusion:
Hepcidin-25 is inversely associated with AKI in a multivariate model when combined with eGFR and diabetes mellitus, with an overall AUC of 0.82. Notably, urinary hepcidin-25 improves on clinical AKI prediction compared to the Thakar score alone.
What was known before
Urinary hepcidin-25 was identified as a novel biomarker of acute kidney injury (AKI) using proteomic techniques 21 , and its diagnostic performance has been evaluated in adult cardiac surgery cohorts.22,37,38 Recently, exogenously administered hepcidin-25 has been shown to be renoprotective in murine models of renal ischemia-reperfusion injury and hemoglobin-mediated AKI that both mimic cardiac surgery–associated AKI.13,26
What this adds
This study validates that urinary hepcidin-25 is a non-invasive early marker for AKI in an independent adult cardiac surgery cohort. This study extends upon the previous work by demonstrating that urinary hepcidin-25 outperforms clinical prediction and serum creatinine.
Introduction
Acute kidney injury (AKI) following cardiac surgery requiring cardiopulmonary bypass (CPB) is a serious complication resulting in increased morbidity and mortality. Mild AKI occurs in 17% of cardiac patients (with CPB), 1 and severe AKI requiring dialysis occurs in 2% of patients with a 60% mortality risk.2-6 Mild renal impairment, defined as a serum creatinine (Cr) >25% from baseline, is associated with a doubling in mortality up to 10 years following cardiac surgery independent of whether renal function recovers to baseline.7,8 Post-operative AKI is also an independent predictor of developing chronic kidney disease.9,10 Non-invasive urinary biomarkers may identify early injury in AKI patients and potentially provide a window for therapeutic intervention prior to loss of function. 11
Hepcidin-25 is a disulfide-rich antimicrobial peptide that regulates circulating iron by degrading and internalizing the cellular iron transporter, ferroportin, on hepatocytes, macrophages, and enterocytes. 12 Renal ischemia reperfusion injury (IRI) is known to increase ferroportin expression in the spleen and liver, inducing hepatosplenic iron export. 13 Tight regulation of ferroportin is necessary to keep circulating iron levels stable. Hepcidin is produced by the liver as an 84-amino acid prepropeptide and is approximately 90% bound to α2-macroglobulin in the circulation. 14 Hepcidin is also expressed in the thick ascending limb, connecting tubules and collecting ducts of the kidney. 15 Cleavage into the bioactive 25-amino acid form 16 with a specific N-terminus is required for ferroportin interaction and intracellular iron sequestration. 17 Hepcidin-20 and -22 isoforms have been detected in human serum and urine, but only hepcidin-25 is associated with ferroportin degradation.18-20
Using an unbiased proteomics approach, we identified urinary hepcidin-25 as a marker for AKI avoidance 21 and demonstrated that it is inversely associated with AKI at postoperative day 1 (POD 1). 22 The goal of this study was to validate its diagnostic performance in an independent cardiac surgery cohort in the context of clinical AKI predictors.
Methods
The study protocol was approved by the University of Manitoba institutional review board (ethics approval HS15221(H2012:097)), and all patients provided written informed consent. All elective coronary artery bypass graft (CABG) and/or valve replacement patients from June 2012-October 2014 were considered for inclusion. Exclusion criteria were off-pump surgeries, pre-existing dialysis dependence or renal transplant, deceased prior to surgery, canceled/delayed surgeries, and missed urine sample bio-banking. Patient demographics and comorbidities were obtained. Intra-operative and post-operative data collection included duration of the operation, CPB and cross-clamp time, urine output, blood pressure, inotropic support, blood products, and potential nephrotoxic exposures (aminoglycoside, non-steroidal anti-inflammatories, angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, and contrast).
Urine Biobanking
Samples were obtained at baseline (start of the operation), during early IRI (start of CPB, 1 hour into CPB), on arrival to intensive care unit (ICU), and POD 1 and 3 to 5 (Figure 1). Urine (20 mL) was collected via a Foley catheter placed after anesthetic induction until ICU discharge; ward patients provided a midstream catch. All samples were immediately placed on ice or stored at 4°C and centrifuged at 870g for 6 minutes. The supernatant was collected and stored at −80°C.

Study design.
AKI KDIGO Criteria
Baseline serum creatinine was established using an average of two preoperative serum creatinine values for all patients. AKI was defined using the 2012 Kidney Disease Improving Global Outcomes (KDIGO) criteria: serum creatinine (Cr) rise >50% from baseline or a ≥26.5 μmol/L serum creatinine increase within any 48 hour period through till POD 4. 23 The KDIGO urine output criterion was not used to determine AKI. 24
Urinary Hepcidin-25 ELISA
Urinary hepcidin-25 concentration was measured with a competitive ELISA (Penlab S-1337) (BioTek Synergy 4 microplate reader, Gen5 software, Fisher Scientific), as previously described. 22 As urine hepcidin-25 has previously been demonstrated to peak at POD 1, these experiments were performed on POD 1 samples.21,22 Briefly, concentrations outside the standard curve range were serially diluted until detection. All samples were run in duplicate with an intra-assay coefficient of variation (CV) of 3.2%. An internal control was included on each ELISA plate using normal urine spiked with hepcidin-25 (20 ng/mL) in order to assess the plate-to-plate variation, or inter-assay CV. The inter-assay CV of two different plate lots was observed to be 0.46% and 23.7%, respectively. Urine creatinine was measured (Roche Cobas 8000 analyzer) to correct for dilutional factors, which is presented as urine hepcidin-25:Cr.
Statistical Analyses
Data were analyzed using SAS version 9.3 (SAS, Cary, North Carolina). Descriptive statistics are expressed as a median (interquartile range) or n (%). The Non-parametric Mann-Whitney test was used to compare continuous variables. Chi-square or Fisher’s exact test was used to compare categorical variables. Multiple imputation was used on missing covariate data. Univariate logistic regression–derived odds ratios (ORs; 95% confidence interval [CI]) were calculated to estimate the association between AKI and all variables, including urine hepcidin-25 concentration and urine hepcidin-25:Cr. The Thakar score is a composite clinical prediction score for predicting AKI following cardiac surgery and includes the following variables: sex, congestive heart failure, left ventricular ejection fraction <35%, preoperative use of intra-aortic balloon pump, chronic obstructive pulmonary disease, insulin-requiring diabetes, previous and other cardiac surgery, emergency surgery, type of cardiac surgery (CABG, valve, both), and baseline renal function. 3
A multivariate logistic regression model was developed for AKI using the stepwise selection method. The primary exposure variable was urinary hepcidin-25 concentration at POD 1. All patient characteristics were considered in building the model. Variables were selected for inclusion based on biological plausibility and statistical significance. Given the high inter-assay CVs observed, we included the internal control as a continuous variable on the final multivariate analysis to control for plate-to-plate variation. Individual ORs (95% CI) were calculated for baseline estimated glomerular filtration rate (eGFR), diabetes mellitus, the internal control, and urine hepcidin-25 concentration. The combined area under the curve (AUC) was determined for receiver operating characteristic (ROC) curves in the final multivariate models. The goodness-of-fit was evaluated using the Hosmer-Lemeshow test. Finally, a supplemental analysis was performed excluding patients that developed AKI on or before POD 1 (n = 6).
Results
Prospective Observational Cohort
Three hundred eighty adult cardiac surgery patients were enrolled in the prospective observational cohort. Twenty-one patients were excluded for off-pump surgeries (n = 3), preexisting dialysis dependence (n = 3), deceased prior to surgery (n = 1), and canceled/delayed surgeries (n = 14). Fifty-three patients were excluded for missed urine sample bio-banking. Therefore, the final prospective observational cohort (n = 306) consisted of 41 AKI and 265 non-AKI patients (Figure 2). The baseline characteristics of patients who were excluded for missing urine samples (n = 53) were compared with the final study population (n = 306), and no significant differences were found between these groups (Supplemental Table 1).

Prospective observational cohort of adult cardiac surgery patients (n = 306).
Predictors of AKI
The patient characteristics of the prospective observational cohort were evaluated by univariate logistic regression in AKI versus non-AKI patients (Table 1). Increased urinary hepcidin-25 [AKI 628 (197-1749) vs. non-AKI 1599 (680-2753)] and urine hepcidin-25:Cr [AKI 67 (29-134) vs. non-AKI 121 (65-208)] was associated with a lower likelihood of developing AKI. Urinary hepcidin-25 (OR 0.61, 95%CI 0.45-0.83, p=0.002) and urine hepcidin-25:Cr (OR 0.65, 95%CI 0.46-0.94, p=0.02) at POD 1 were both univariate predictors for the subsequent development of AKI Additional univariate predictors for AKI development were baseline renal function as determined by eGFR [OR 0.95, 95%CI 0.94-0.97, p<0.0001] and serum creatinine [OR 1.02, 95%CI 1.01-1.03, p<0.0001]; Thakar score [OR 1.63, 95% CI 1.35-1.97, p<0.0001]; diabetes [OR 3.82, 95%CI 1.93-7.54, p<0.0001] and a previous history of hospitalized congestive heart failure [OR 3.82. 95%CI 1.72-8.47, p=0.0005].
Study Population Characteristics.

Note. Values expressed as median (interquartile range) or n (%). Continuous variables compared using Mann-Whitney test, categorical variables compared using chi-square or Fisher’s exact test. Missing covariate values were estimated via multiple imputation. eGFR, estimated glomerular filtration rate; CABG, coronary artery bypass graft; Cr, creatinine.
Odds ratio: aAge (per year); bTime (per hour); cIntraoperative urine output (per L); dUrine hepcidin-25 (per 1000 ng/mL); eUrine hepcidin-25:Cr (per 100 μg/mmol).
Urinary hepcidin-25 was inversely associated with AKI avoidance (OR: 0.67, 95% CI: 0.50-0.95, P = .02) on multivariate analysis, with higher concentrations associated with a lower likelihood of developing AKI (Table 2). A combined multivariate model was developed using baseline eGFR, diabetes mellitus, and urinary hepcidin-25. This model demonstrated AKI prediction with an AUC of 0.82 (95% CI: 0.75-0.88) and reasonable goodness-of-fit (Hosmer-Lemeshow, P = .90) (Table 2).
Urinary Hepcidin-25 is Independently Associated with AKI on Multivariate Analysis.
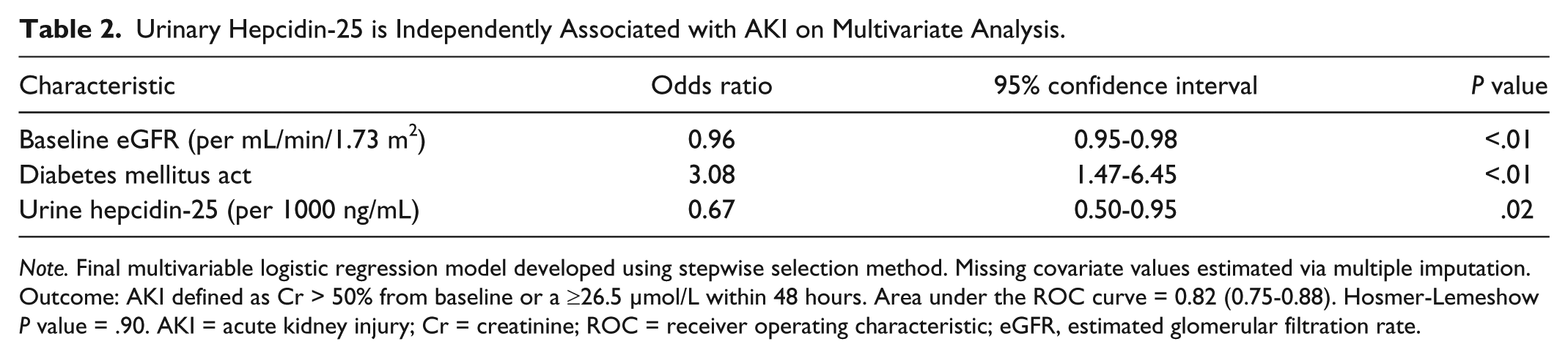
Note. Final multivariable logistic regression model developed using stepwise selection method. Missing covariate values estimated via multiple imputation. Outcome: AKI defined as Cr > 50% from baseline or a ≥26.5 μmol/L within 48 hours. Area under the ROC curve = 0.82 (0.75-0.88). Hosmer-Lemeshow P value = .90. AKI = acute kidney injury; Cr = creatinine; ROC = receiver operating characteristic; eGFR, estimated glomerular filtration rate.
In order to control for the observed plate-to-plate variability, the multivariate analysis was recapitulated including the internal control as a variable. Urinary hepcidin-25 remained a predictor of AKI (OR: 0.70, 95% CI: 0.51-0.96, P = .03). The combined AKI prediction model using baseline eGFR, diabetes mellitus, and urinary hepcidin-25 demonstrated an AUC of 0.83 (95% CI: 0.76-0.89) and reasonable goodness-of-fit (Hosmer-Lemeshow, P = 0.72) (Table 3). The Thakar score was not significant in either analysis.
Urinary Hepcidin-25 is is Inversely and Independently Associated with AKI, After Controlling for Plate–to-Plate Variability.

Note. Final multivariable logistic regression model developed using stepwise selection method. Missing covariate values estimated via multiple imputation. Outcome: AKI defined as Cr > 50% from baseline or a ≥26.5 μmol/L within 48 hours. Area under the ROC curve = 0.83 (0.76-0.89). Hosmer-Lemeshow P value = .72. AKI = acute kidney injury; Cr = creatinine; ROC = receiver operating characteristic; eGFR, estimated glomerular filtration rate.
Supplementary Analyses
To explore if urinary hepcidin-25 is an early predictor of AKI, we excluded patients who developed AKI on or before POD 1 (n = 6), and repeated the multivariate analysis (n = 300, prospective observational cohort). Urinary hepcidin-25 remained an independent predictor for AKI (OR: 0.64, 95% CI: 0.44-0.95, P = 0.03) in the revised model. Furthermore, the combined clinical predictor and biomarker model with baseline eGFR, diabetes mellitus, urinary hepcidin-25, and the internal control still showed strong AKI prediction with an AUC of 0.84 (0.77-0.91). The internal control for plate-to-plate variability was not statistically significant in this model (P = .11) (Table 4). Finally, urine hepcidin-25:Cr was not significant on multivariate analysis (n = 306 population, (OR: 0.77, 95% CI: 0.53-1.12, P = 0.16) and n = 300 population (OR: 0.74, 95% CI: 0.49-1.14, P = 0.18), after correcting for dilutional factors.
Urinary Hepcidin-25 is Inversely and Independently Associated with AKI, After Excluding Patients That Developed AKI on or Before Post-operative Day 1.

Note. Final multivariable logistic regression model developed using stepwise selection method. Missing covariate values estimated via multiple imputation. Outcome: AKI defined as Cr > 50% from baseline or 26.5 μmol/L in 48 hours. Area under the ROC curve = 0.84 (0.77-0.91). Hosmer-LemeshowP value = .46. AKI = acute kidney injury; Cr = creatinine; ROC = receiver operating characteristic; eGFR, estimated glomerular filtration rate.
Discussion
The principle finding of this study was that urinary hepcidin-25 is inversely associated with AKI avoidance in an independent, prospective observational adult cardiac surgery cohort. Importantly, a combined clinical and biomarker model with baseline eGFR, diabetes and hepcidin-25 demonstrated an AUC of 0.82 for the subsequent development of AKI. These findings are consistent with our previous study 22 and extend them by demonstrating that they outperformed clinical AKI prediction with the Thakar score.
All cardiac surgery patients are exposed to IRI with varying degrees of tubular stress or injury, 25 resulting in activation of different renoprotective mechanisms. As urinary hepcidin-25 is increased in non-AKI versus AKI patients following IRI, we previously postulated that it is a renoprotective response which may be mediated via the intracellular sequestration of iron to limit oxidative stress, free radical damage, and renal injury. 21 Since then, Scindia et al have found hepcidin to be highly protective in a murine model of renal IRI. Notably, the administration of hepcidin resulted in a significant reduction in tubular injury, apoptosis, renal oxidative stress and neutrophil infiltration, as well as improved renal function. 13 Furthermore, these findings are highly consistent with van Swelm et al who demonstrated that the administration of hepcidin-25 decreased markers of kidney injury in a murine model of hemoglobin-mediated AKI. 26
Labile ferrous iron release is an inevitable consequence of cardiac surgery. The mechanical forces exerted during extracorporeal circulation induces hemolysis and free hemoglobin release, 27 with increasing CPB duration correlated with increased free hemoglobin release. 28 Elevated free hemoglobin levels are associated with AKI following on-pump repair of aortic aneurysms. 25 Red blood cell (RBC) storage can result in loss of structural integrity and increased hemolysis following blood transfusion,29,30 while myoglobin release may also contribute to the circulating labile iron pool. 31 Finally, renal IRI alone can induce increased serum iron levels, with associated kidney iron accumulation. 13 The intersection of these findings with the observed renoprotective effect of hepcidin in murine models of renal IRI and hemoglobin-mediated AKI, all suggest that iron handling is a crucial and potentially modifiable factor of cardiac surgery–associated AKI.
While hepcidin-25 is renoprotective in animal models, its underlying mechanisms in cardiac surgery–associated AKI remains unknown as there are conflicting observations regarding the role of apoptosis and renal ferroportin.13,26 Notably, these observations suggest that hepcidin-25 may act via alternative pathways to prevent renal tubular epithelial cell death.13,26 Ferroptosis is a novel iron-dependent form of regulated cell death that has recently been demonstrated to play a key role in AKI-induced cell death.32,33 Indeed, inhibition of ferroptosis has added protective effects beyond necroptosis and necrosis inhibition alone in a murine model of severe renal IRI. 32 Furthermore, ferroptosis was the primary mediator of cell death and renal injury in folic acid–induced AKI. In this model, inhibition of ferroptosis protected renal function, histological injury, and cell death, whereas inhibition of apoptosis and necroptosis were ineffective. 33 These findings are intriguing in that they delineate an alternative iron-dependent pathway to renal injury that is shared between different types of AKI. It is tempting to speculate that the lack of consistently observed inhibition of apoptosis by hepcidin-25 administration13,26 may be due to activation of alternative cell death pathways, such as ferroptosis, but this cannot be clarified in our human model of IRI-AKI. These findings are consistent with data on another iron-binding protein, neutrophil gelatinase-associated lipocalin (NGAL), which has been demonstrated to be renoprotective as part of an NGAL:iron:siderophore complex that upregulates heme-oxygenase. 34 Taken together, these findings suggest that hepcidin-25 may be part of a coordinate response involving different iron-binding proteins that act to mitigate the response to human renal IRI.
Hepcidin is freely filtered by the glomerulus and reabsorbed at the proximal tubule via megalin-dependent endocytosis26,35; the fractional excretion is negligible with up to 97% reabsorbed in physiological conditions. 36 While the source of the observed urinary hepcidin-25 in our human model is unknown, hepcidin is locally synthesized in kidney and released in the distal nephron in AKI. 26 These findings are consistent with cardiac surgery models of AKI which demonstrated increased urinary hepcidin-25 and increased fractional excretion of hepcidin-25 in non-AKI patients, in the absence of substantive changes to plasma/serum hepcidin-25, even after excluding chronic kidney disease patients.37-38 Taken together, we speculate that elevation of urinary hepcidin-25 following cardiac surgery is the result of increased distal nephron production in response to IRI, but it remains to be determined whether luminal enzyme activity plays an in vivo regulatory role by inactivating hepcidin-25.
This study had several limitations. First, this is strictly an observational study. Although we speculate that urinary hepcidin-25 is elevated in human IRI as part of a coordinated renoprotective response, causation cannot be determined. Second, this is a single-center study. These findings confirm those of other single-center studies,22,37,38 although urine hepcidin:Cr was not significant after correcting for dilutional factors in this study. It is possible that the urinary hepcidin-25 concentration in the lumen of the distal tubule is predictive of protection, instead of mass excretion rate. An alternative possibility is that urinary hepcidin-25 may be less sensitive for milder phenotypes of AKI, as this cohort consisted of largely Stage 1 AKI (Stage 1, n=38; Stage 2, n=1; and Stage 3, n=2). Therefore, these findings need to be verified in a multi-center setting. The patterns of hepcidin-25 excretion in other human AKI models remains unknown; however, the observed effect of iron-dependent cell death in different mouse AKI models suggests that iron may be a key regulator to cell death/injury beyond IRI or hemoglobin-mediated AKI. Fifty-three patients were excluded for missed bio-banking, although no significant differences were identified between the excluded and final study population.
Hepcidin-25 is limited as an early, non-invasive biomarker by its polyclonal antibody–based ELISA assay. This resulted in significant plate-to-plate variation between lots, which we identified with our internal controls. We accounted for this variability in the analysis and demonstrated that it did not substantially impact the overall findings. While a hepcidin-25 monoclonal antibody sandwich ELISA has been developed, 39 these antibodies were not available at the time of our experiments. Clinically applicable biomarkers require a robust, reproducible assay that is readily translatable to clinical laboratories and conform to Good Laboratory Practice guidelines.40,41 Finally, hepcidin-25 peaks later at POD 1, similar to KIM-1 (kidney injury molecule-1), and therefore has limited utility for early diagnosis compared with biomarkers that peak earlier. Nevertheless, this cohort provides useful independent observations regarding the consistent response of urinary hepcidin-25 as a marker of AKI avoidance and helps shed insight into the pathophysiology of human renal IRI.
Conclusions
Urinary hepcidin-25 is elevated following cardiac surgery and is inversely associated with AKI on an independent level. These findings independently validate the existing literature in a prospective, observational cohort of adult cardiac surgery patients,21,22,37,38 and extend them to show that a combined clinical and biomarker model with baseline eGFR, diabetes mellitus, and hepcidin-25 has an AUC of 0.82 for the subsequent development of AKI. These findings suggest that iron-dependent pathways are key mediators of renal IRI and are consistent with animal models that demonstrate hepcidin-25 is renoprotective in AKI. It remains to be determined whether iron modulation can mitigate the effects of human AKI, and the results of a clinical trial evaluating the utility of iron chelation in AKI with deferoxamine are pending (NCT00870883).
Footnotes
Acknowledgements
The authors gratefully acknowledge the Adult Cardiac Surgery Program at St. Boniface Hospital and the operating room staff for their assistance with the collection of this cohort and Evelyn Roloff for her assistance with preparation of the manuscript.
Ethics Approval and Consent to Participate
The study protocol was approved by the University of Manitoba institutional review board, and all patients provided written informed consent (ethics approval HS15221 (H2012:097).
Consent for Publication
We have authors consent for publication.
Availability of Data and Materials
Data is available upon request.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The prospective observational cohort of adult cardiac surgery patients was collected with funding support by Research Manitoba (J.H.). The urine hepcidin-25 analysis was funded by the Kidney Foundation of Canada (J.H.) and Christella I Cann award (J.H., M.Z.). J.H. holds a Canadian Institutes of Health Research New Investigator Salary Award.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.