
Abstract
The development of vaccines that target tumor antigens in cancer has proven difficult. A major reason for this is that T cells specific for tumor self-antigens and neoantigens are eliminated or inactivated through mechanisms of tolerance. Antigen fusion strategies which increase the ability of vaccines to stimulate T cells that have escaped tolerance mechanisms, may have a particular potential as immunotherapies. This review highlights antigen fusion strategies that have been successful in stimulating the induction of T-cell immunity against cancer and counteracting tumor-associated tolerance. In preclinical studies, these strategies have shown to improve the potency of vectored vaccines through fusion of tumor antigen to proteins or protein domains that increase CD4+ T-cell help, CD8+ T-cell responses or both the CD4+ and CD8+ T-cell responses. However, in clinical trials such strategies seem to be less efficient when provided as a DNA vaccine. The first clinical trial using a viral vectored fusion-gene vaccine is expected to be tested as a partner in a heterologous prime-boost regimen directed against cervical cancer.
Keywords
Introduction
Cancer is one of the major causes of mortality worldwide. Therefore, a great deal of effort has been put into developing vaccines to prevent or treat the disease. Whilst the anti-cancer potential of prophylactic vaccines has already been proven with the Hepatitis B vaccine, it has not yet been possible to induce regression of existing tumors by therapeutic vaccination. Clearly, the development of functional cancer vaccines has proven much more difficult than vaccination against infectious diseases. A major reason for this difficulty is that tumors arise from the individual’s own tissue and only the few genes that are mutated express proteins that will be seen as foreign by the immune system. Since tumors are extremely heterogenous and generally arise by casual mutations, the development of therapeutic vaccines targeting different cancers represents a difficult challenge. In addition, up-regulation and over-expression of self-antigens in tumorgenesis do not necessarily induce a functional immune response. This is because T cells reacting to such antigens have been negatively selected within the thymus or eliminated by the induction of tolerance in the periphery [Xing et al., 2012]. In addition, cancer cells may present insufficient stimulation for full activation of professional antigen presenting cells (pAPCs). This leads to insufficient expression of major histocompatibility complex class I and II-peptide molecules (MHC-I and MHC-II), co-stimulatory molecules and cytokine production, which via a similar mechanism of induction of peripheral tolerance, may induce anergy in T cells reactive against growing cancers [Hawiger et al., 2001]. Moreover, the thymic selection processes, peripheral tolerance and tumor antigen presentation, lead to the induction of regulatory T cells (Tregs) with specificity for self and tumor-associated antigens [Bilate et al., 2012]. Once generated and expanded during tumor progression, these cells will seek to dampen subsequent immune responses against self and tumor antigens. Collectively, these factors explain why T cells recognizing tumor-associated antigens (TAAs) are usually present at very low frequencies and are often not very responsive to stimulation. Furthermore, even when tumor reactive T cells are generated, cancer cells create a locally immune suppressive environment by down-regulating the expression of MHC-I molecules on their surface, releasing immunosuppressive cytokines and enhancing the expansion of Tregs as well as myeloid derived suppressor cells (MDSCs) [Mellman et al., 2011]. Taken together, these aspects prevent the generation of immune effectors, such as CD4+ T helper or CD8+ cytotoxic effector T cells, or limit the efficiency of responses that are raised.
A prerequisite for induction of efficient antitumor responses is the “breaking” of T-cell unresponsiveness towards cancer cells, as well as the neutralization of local tumor-associated immune suppressing mechanisms. This review describes how genetic strategies, which increase antigen presentation by coupling vector-delivered antigens to cis acting adjuvants, can break T-cell unresponsiveness towards tumors and improve the efficacy of cancer vaccines.
Overcoming T-cell tolerance
T-cell tolerance
Tolerance to a given antigen is acquired by either induction of central tolerance in the thymus or by induction of peripheral tolerance extrathymically.
During thymic selection high-affinity self reactive clones are eliminated by clonal deletion [Xing et al., 2012]. Approximately 50-60% of positively selected thymic T cells are eliminated in this process [Ignatowicz et al., 1996, Van Meerwijk et al., 1997]. It has been hotly debated whether all self-antigens are expressed in the thymus. It is logical that the thymic medullary epithelial cells cannot express all antigens to the levels that are seen in highly specialized organs dedicated to maximal production of a small number of proteins (e.g. insulin in the endocrine pancreas, thyroglobulin in the thyroid gland). Therefore, thymic deletion is bound to be incomplete, with the consequence of possible escape of auto-reactive T cells entering the periphery where they must be controlled by peripheral tolerance. An important mechanism of peripheral tolerance is the continuous antigen sampling by dendritic cells (DCs) in the absence of inflammatory stimuli. This leads to antigen display in the absence of co-stimulation followed by deletion or inactivation of auto-reactive T cells via clonal elimination, clonal diversion, receptor editing and anergy [Klein et al., 1998, Kyewski et al., 2002]. Although both central and peripheral tolerance in most cases is sufficient to prevent autoimmunity, the mechanisms are intrinsically incomplete. Antigens sampled by DCs can only be presented to CD8+ T cells via cross-presentation, which rarely leads to antigen presentation at levels similar to the target cell synthesizing the antigen endogenously [Ochsenbein et al., 2001]. This incomplete selection induces auto-reactive T cells that have an avidity of antigen recognition sufficient to target specific tissues but insufficient to be targeted by central and peripheral tolerogenic mechanisms. These cells are prevented from causing disease, principally because such naïve T cells are absent in the peripheral tissues and because the antigen is insufficiently displayed in lymph nodes to support their clonal expansion and maintenance. However, the incomplete elimination of self-reactive cells still has significant consequences. This can be demonstrated when viral epitopes mimic self-epitopes and cause auto-immunity after triggering of self-reactive T cells. It has been speculated that this is the case for some chronic autoimmune diseases such as multiple sclerosis, where chronic viral infection with Epstein Barr Virus triggers self-reactive T cells [Lang et al., 2002]. Similarly, it is thought that the mumps virus can trigger acute autoimmune disease orchitis [Schuppe et al., 2008].
Importantly, even though numerous check-points are established to prevent tissue reactivity in the first place, additional mechanisms are also available to reduce inflammation once established. In this context, natural and inducible Tregs play an important role due to their sensitivity to inflammation and T-cell responses via expression of the high affinity IL-2 receptor [Almeida et al., 2002, Hofer et al., 2012]. Tregs can expand during cancer progression and are present in the tissue and particularly within tumors ahead of the T-cell response. Both in tumor draining lymph nodes and within tumors, Tregs accumulate [Schneider et al., 2011] and are believed to exert a powerful suppression of the effector phase of the T-cell response. As Tregs per definition can suppress immune responses, most studies suggest a negative correlation between tumor infiltration by Tregs and long-term survival [Shen et al., 2010]. Conversely, in some cancers a strong positive correlation between Tregs and survival is found, quite possibly because the magnitude of CD8+ T-cell responses is co-variable with Treg activity [West et al., 2012]. Furthermore, MDSCs are expanded in chronic inflammatory settings and can create strongly tolerogenic environments, particularly within tumors but also in tumor draining lymph nodes and systemically [Marigo et al., 2008]. The mechanism which enables Tregs and MDSCs to determine when inflammation should be allowed or abrogated are disputed and they may not truly exist [Zinkernagel et al., 2004]. Possibly, they will simply regulate all inflammation and will be dominating once the offending signal becomes weaker. Nevertheless, together with homeostatic tolerogenic mechanisms, Tregs and MDSCs pose a formidable obstacle for cancer immunotherapy which must be overpowered to achieve anticancer efficacy by vaccination.
Even though tumors arise from the organism’s own tissue, they will eventually become antigenic either through abnormal gene expression patterns or by mutations which create genuine neo-antigens, and the immune system targets such antigenic specificities [Urban et al., 1992]. Evidence of this can be seen as tumors grown in immunocompetent mice are less immunogenic than similar tumors raised in immunodeficient mice [Dunn et al., 2004]. As it turns out, tumors apply mechanisms similar to peripheral tolerance by cross-presentation of tumor antigens by bone-marrow- derived APCs. This APC-T cell encounter can result in loss of T-cell clonal expansion as well as partial activation, followed by unresponsiveness of T cells [Sotomayor et al., 2001]. A second major mechanism is the deletion of tumor antigen-reactive T cells within the tumors. Thus, longitudinal sampling of tumors and clone specific mapping of infiltrating T cells reveal that primed tumor-specific T cells have a short lifespan within the tumors and do not reoccur [Thor Straten et al., 2004]. A range of factors have been implicated in this T-cell killing. These include depletion of tryptophan by local up-regulation of idoleamine 2,3-dioxygenase (IDO) expression and expression of apoptosis-inducing factors such as Fas ligand or B7-H1 on the surface of tumor cells [Strand et al., 1996, Walker et al., 1998, Dong et al., 2002, Platten et al., 2012]. A rather profound point is that this induction of tumor- specific T-cell anergy has been shown to occur at the early stages of tumor progression [Overwijk et al., 1998]. Taken together, this data indicates that only a low number of tumor-specific T cells are induced at any particular time, which leads to inefficient control of tumor-progression and either deletion of the clone or, probably less frequently, selection of escape mutations in the tumor [Dunn et al., 2002]. Thus, in any particular tumor bearing host the immune system has already been specifically educated to allow progression of the tumor and importantly this would also hold true for tumors promoted by genuine neo-antigens and those driven by viral infection. This “immune education” will not always be manifested in tumor experiments using transplantable cell lines. Limited education of the T- cell repertoire is, together with a dramatic tumor heterogeneity, suggested to be a major cause of discrepancy between many pre-clinical and clinical cancer immunotherapy trials. Fortunately, like the tolerogenic mechanisms preventing autoimmune diseases, tolerance is incomplete, but to counteract these selection principles, vaccination must overpower the suppressive mechanisms. This could in principle be done by inducing a high number of tumor reactive T cells at one time point or alternatively, by promoting continuous T-cell expansion. Naturally, combination treatments which employ inhibition of tumor-specific mechanisms of T-cell inhibition would be predicted to be much more efficient [Sorensen et al., 2010]. Although we have yet to see a vaccine concept which can massively expand tumor-specific T-cells in vivo, the general concept of combating tumors with very high numbers of T cells has found strong, if indirect, experimental support. Thus, harvesting of tumor infiltrating lymphocytes (TIL) from metastatic melanomas, followed by their ex vivo expansion and re-infusion into the patients result in dramatic and frequently curative anti-tumor efficacy [Yee et al., 2002]. Theoretically, potent vaccines should be able to do the same. The mechanisms of central tolerance, peripheral tolerance, cancer-induced T-cell tolerance and activation of low avidity T-cells are presented in figure 1.

Tolerance and cancer. Panel A represents the negative selection involved in central tolerance, which takes place in the thymus. This mechanisms consist in deleting self-reactive T-cell clones that recognized with a high affinity self-antigen expressing on the surface of immature dendritic cells (iDC). Panel B represents the peripheral tolerance in lymph nodes, here exemplified in the pancreas. Autoreactive T cells that escape selection in the thymus are then controlled by peripheral tolerance. iDCs sample tissue antigens displays the self peptides to T cells. The absence of co-stimulation lead to deletion or inactivation of autoreactive T cells. Panel C represents the mechanism of “tolerance” induced by tumor cells. Tumor cells induce insufficient danger signals to fully activate DCs. The effects induced to the tumor-specific T cells are similar as those explained in Panel B. Panel D represents the activation of low avidity autoreactive T cells by mature DCs, which can target and kill tumor cells. These low avidity T cells have escaped central and peripheral tolerance; their activation requires high epitope density as well as co-stimulation. TAA, tumor-associated antigen; DCs, dendritic cells; iDCs, immature dendritic cells, mDCs, mature dendritic cells; MHC, major histocompatibily complex.
Breaking tolerance
Although central and peripheral tolerance are efficient in eliminating high avidity auto-reactive T cells, low avidity T cells can escape these mechanisms [Von Herrath et al., 1994a]. Under certain conditions these low avidity T cells can be functional and cause autoimmune disease. As an example, Von Herrath and colleagues studied tolerance mechanisms using transgenic mice in which pancreatic β-cells expressed the glycoprotein of Lymphocytic choriomeningitis virus (GP of LCMV) under control of the rat insulin promoter (RIP). This transgenic GP expression did not cause insulin-dependent diabetes mellitus (IDDM). However, LCMV infection in these transgenic mice resulted in an immune response to the β-cells and progression to IDDM. In this model, self- specific CD8+ T cells were of lower affinity and avidity than CD8+ T cells generated by LCMV infection in the nontransgenic control [Von Herrath et al., 1994b]. Few studies have analyzed if these residual T cells of lower affinity can be exploited for cancer therapy. In 2010, Sorensen and colleagues tested a new vaccine, which consisted of a replication deficient adenovirus expressing the murine MHC-II invariant chain linked to the GP of LCMV (Ad-IiGP). The invariant chain (Ii) fusion increases the antigen specific cell surface presentation of the MHC/peptide complex on the surface of APCs and the vector provides up-regulation of costimulatory molecules. The efficient antigen presentation was correlated with a stronger T-cell response in wild type (WT) mice [Holst et al., 2008]. Next, they investigated if this vaccine would be efficient against tumors expressing an endogenous antigen. For this purpose, they used the RIP expressing GP transgenic C57BL/6 mice mentioned above. Transgenic and nontransgenic mice were then challenged with B16.F10 melanoma cells expressing the MHC-I restricted immunodominant epitope of LCMV (GP33), followed a few days later by immunization with either Ad-IiGP or Ad-GP. In nonstrangenic tumor bearing mice the Ad-GP vaccine and to a larger extent the Ad-IiGP vaccine delayed the tumor growth compared to unvaccinated mice. However, in transgenic tumor bearing mice, the conventional vaccine expressing GP had no effect on the tumor progression while the Ad-IiGP vaccine was equally protective in nontransgenic tumor bearing mice [Sorensen et al., 2009]. Taken together, these data suggest that activation of low avidity T cells requires high MHC-I/II restricted epitope density as well as costimulation to induce tumor rejection. However, once triggered by an efficient vaccine these cells are able to kill tumor cells, proving that central, peripheral and tumor-specific tolerance is incomplete and can be counteracted by fusion-antigen vaccines. It should be noted that low avidity T cells are not suggested to be uniquely beneficial in anti-cancer immunity. Fusion-antigen vaccines will select for high and low avidity T cells alike and their theoretical potential stems from an improved ability to stimulate the T cells that are available. Once generated, the T cells with low affinity or avidity as such may have a particular survival advantage in tumor bearing hosts [Caserta et al., 2010], but in the effector phase of a response the avidity is a definite positive correlate of efficacy as can be demonstrated by direct alteration of T-cell responsiveness without altering specificity [Stromnes et al., 2010]. A more pronounced theoretical advantage of low affinity T cells stems from the higher immunogenicity of tumors than non-inflamed tissues. Thus, as above-mentioned, low affinity T cells can remain in a naïve state but be triggered for anergy by tumor progression and tumor immunity by infection [Lyman et al., 2005]. Such findings suggest that tumors that are to be controlled by low avidity T cells will not later suffer from autoimmune diseases. Anecdotal evidence for this has been seen in humans where IL-2 treatments sometimes result in tumor rejection. In a particularly well studied patient, persistence of survivin-specific T cells were observed for years after apparent tumor clearance without evidence of autoimmune disease [Hadrup et al., 2006].
In line with this data, a way of breaking T-cell tolerance towards cancer cells will be to increase antigen presentation. Alternative strategies, such as increasing costimulation, addition of cytokines and epitope optimization are not the subject of this review. The readers are referred to other reviews on the topic [Holst et al., 2010, Berzofsky et al., 2012]. Increasing antigen presentation can be done by direct linkage in cis of the antigenic sequence to another sequence that acts to enhance the antigen presentation.
Innovative vector vaccines breaking immune tolerance against cancer: cis-acting fusion gene vaccines
Difference between DNA and viral-vectored vaccines
Most improvements to increase the anti-tumor immune responses have been investigated using DNA vaccines and to a lesser extent using viral vectored vaccines. DNA vaccines are appealing because of their safety, simplicity and low cost. A DNA vaccine simply requires the cloning of a plasmid with an expression cassette and an antigen-encoding sequence. However, they are poorly immunogenic which results in insufficient efficacy. To solve this problem, cis-acting sequences have been used and showed great improvements, notably against cancer as discussed below. A more generally applicable approach has been to increase tissue transduction of DNA vaccines by electroporation. Electroporation increases tissue transduction but also provides an inflammatory signal which together with the increased transduction may augment the immune response [Liu et al., 2008]. Although doubts have been raised about the clinical prospects of electroporation in humans due to the quite easily felt electrical shock, recent clinical trials indicate that the procedure is well tolerated and importantly, effective [Vasan et al., 2011].
Compared to regular DNA vaccines, viral-vectored vaccines are frequently highly immunogenic without any sequence modifications. Furthermore, compared to a natural infection or vaccination with a live-attenuated vaccine, they offer an improved safety profile. They act as “viruses” and efficiently transduce cells, including pAPCs, and they also induce high levels of protein expression in the transduced cells. Moreover, viral vectors contain viral proteins that are immunogenic. As a result, high amounts of antigen are immediately available to stimulate both CD4+ and CD8+ T cells against vector-derived proteins and consequently, CD4+ T helper cells are available during the priming of the vaccine antigen specific precursor T cells. A downside of viral vectored vaccines is that vector antigens compete with the encoded vaccine antigen for stimulation of CD8+ T cells. Subsequently, this focuses the ensuing response on immunodominant epitopes in the antigen [Schirmbeck et al., 2008]. However, this problem is principally solvable by using helper-dependent viral vectors which do not express any viral genes [Kron et al., 2011]. At the injection site, transduced cells produce a substantial amount of antigens that are available for cross-presentation and in fact this is the quantitatively most important source of antigen for induction of antigen specific CD8+ T cells, at least by regular viral vectors not expressing fusion antigens [Prasad et al., 2001]. Although viral vectors have other limitations, it is evident that viral vectors are superior to standard DNA vaccines in most of the qualities that may be improved by antigen coupling strategies [Bett et al., 2010]. DNA vaccines with electroporation might constitute a separate entity with high potency that may even surpass single adenovirus vaccines after repeated vaccinations [Hirao et al., 2010]. Comparisons between DNA administered with electroporation and viral vectors delivered in prime-boost immunizations are still lacking.
In the following section we will discuss the different improvements in vector vaccines (listed in table1) against cancers incorporating fusion-genes and the clinical trials that have been initiated with these vaccines. The different strategies and their effect on T-cell responses are represented in figure 2.
Different strategies of vectored gene fusion vaccines that enhance anti-tumor immune responses
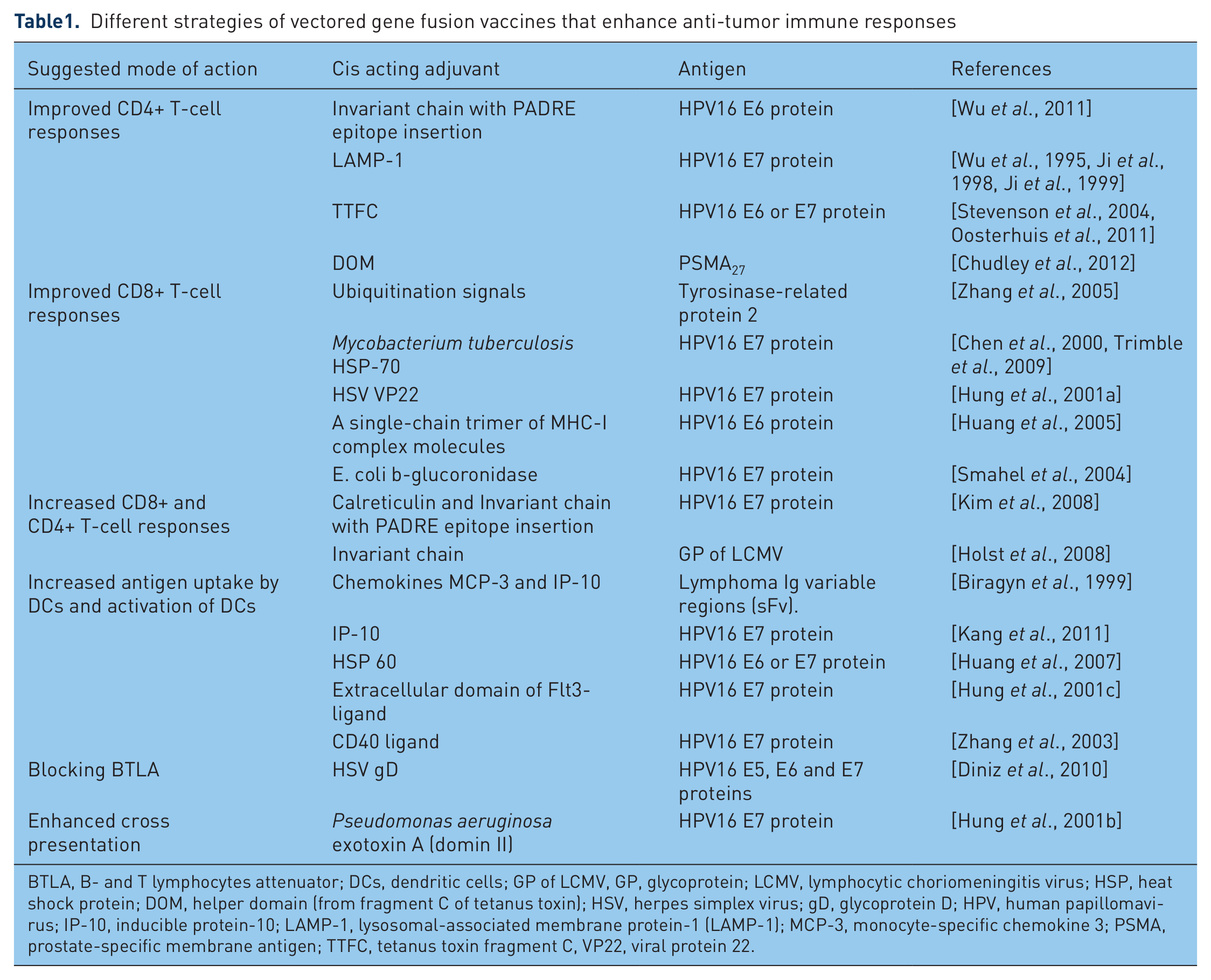
BTLA, B- and T lymphocytes attenuator; DCs, dendritic cells; GP of LCMV, GP, glycoprotein; LCMV, lymphocytic choriomeningitis virus; HSP, heat shock protein; DOM, helper domain (from fragment C of tetanus toxin); HSV, herpes simplex virus; gD, glycoprotein D; HPV, human papillomavirus; IP-10, inducible protein-10; LAMP-1, lysosomal-associated membrane protein-1 (LAMP-1); MCP-3, monocyte-specific chemokine 3; PSMA, prostate-specific membrane antigen; TTFC, tetanus toxin fragment C, VP22, viral protein 22.

Strategies for breaking tolerance using vector fusion-gene vaccines. The three panels represent different strategies of vaccination targeting dendritic cells (DCs) in order to stimulate tumor-specific T cells. Yellow arrows in the DC highlight well-established sorting pathways for MHC-I and II molecules. On the left side of the DC, yellow arrows show the endocytic MHC-II loading compartments whilst on the right side they show the endogenous MHC-I loading compartments. Panel A describes the effect of a vector vaccine expressing tumor-associated antigen (TAA) only. This strategy is here depicted as inducing insufficient expression of tumor antigen-MHC-I complex on the surface of DC to efficiently activate the low affinity tumor-specific CD8+T cells available. Panel B illustrates the enhancement of CD4+ T-cell responses using vector vaccines encoding tumor antigen fused to endosomal sorting motifs (LAMP-1/li) or broadly recognized T helper epitopes. Activated CD4+ T cells then produce cytokines to help cytotoxic CD8+ T cells grow and differentiate. Panel C represents the improvement of CD8+ T cells response using vector vaccines. Most of them target the endogenous MHC-I loading compartment (right side of DC), and improve the loading of antigen to the MHC-I complex, which induce activation and expansion of tumor-specific CD8+ T cells. li and VP22 are the exceptions and a mechanistic understanding of their mechanism of action is still not available although li is indeed an ER chaperone.
Strategies improving CD4+ T-cell responses to help tumor-specific CD8+ T-cell responses
Many vaccine strategies are focused on enhancing tumor-specific cytotoxic CD8+ T cells. However, CD4+ T lymphocytes play a central role in regulating anti-tumor immunity. Indeed, they have the capacity to help cytotoxic CD8+ T cells by producing cytokines that facilitate their growth and differentiation but they can also directly or indirectly kill tumor cells. In addition, they are a key element in inducing long-term immune memory [Ostrand-Rosenberg, 2005]. A successful strategy to increase DNA vaccine-induced CD4+ T-cell responses has been made by including an irrelevant but broadly recognized T (Th)- cell epitope or Th epitope containing protein, such as PADRE [Wu et al., 2011]. DNA vaccine-induced CD4+ T-cell responses have also been improved by targeting the antigen into the MHC-II loading compartment. For example, in a mouse model the administration of a DNA vaccine expressing the E7 protein of the human papillomavirus 16 (HPV 16) linked to the lysosomal-associated membrane protein 1 (LAMP-1) enhanced the activation of tumor- specific CD4+ T cells and increased the number of tumor-specific CD8+ T cells. Immunized mice were significantly protected against E7 positive expressing tumors and metastatic disease [Wu et al., 1995, Ji et al., 1998, Ji et al., 1999].
Moreover, in a nontumor model this strategy was shown to influence the phenotype of T cells. It shifted from a central-memory to an effector-memory phenotype compared with the administration of unlinked antigen [Valentin et al., 2009].
With regard to viral vectors, a novel vaccination strategy has shown to improve both the antigen specific CD4+ T-cell response and the neutralizing antibody response. The strategy consists of an adenoviral vector that encodes and displays a vaccine antigen on the capsid. The antigen, which is the envelope protein gp70 of Friend murine leukemia virus (FV), is fused to the adenovirus capsid protein IX. This approach leads to a significantly improved protection against FV infection in comparison to vaccination with conventional adenoviral vectors [Bayer et al., 2010]. Unfortunately, it has not yet been tested in a tumor model.
Strategies specifically enhancing tumor-specific CD8+ T cells responses
As previously discussed, the density of the tumor antigen-MHC class I complex on the cell surface may influence the cytotoxic CD8+ T-cell response towards tumor cells. For this purpose many strategies have been devised to improve the loading of antigen to the MHC-I complex, including the addition of ubiquitination signals to the antigen, direct coupling of antigen to a single-chain trimer of MHC-I complex molecules, fusion of antigen with chaperones or by increasing intercellular spreading with viral protein-derived transduction domains [Chen et al., 2000, Hung et al., 2001a, Huang et al., 2005, Zhang et al., 2005]. These strategies have been relatively successful in increasing antigen presentation leading to a strong tumor-specific CD8+ T-cell response and interestingly, different molecular mechanisms seem to be involved in the increase of antigen presentation. As a first example, Zhang and colleagues described the use of a naked DNA vaccine encoding a fusion protein linking murine ubiquitin (UB) to the N-terminus of a full-length self antigen. This strategy is based on the rapid destruction of cellular proteins through the UB fusion degradation (UFD) pathway [Zhang et al., 2005]. Ubiquitination signals may act to increase proteasomal degradation and therefore, transport associated with antigen processing (TAP)-mediated transport into the endoplasmic reticulum (ER) for MHC-I antigen loading. This leads to a strong CD8+ T-cell response and a protective immunity to melanoma in mice [Zhang et al., 2005]. The second example is the use of a naked DNA vaccine encoding a tumor antigen linked to beta-2-microglobulin (β2-microglobulin) which is noncovalently linked to the MHC-I heavy chain. This linkage is believed to function by presentation of an intact MHC-I/β2-microglobulin/antigenic peptide heterotrimer on the cell surface, thereby bypassing normal MHC-I antigen processing pathways [Huang et al., 2005]. A major limitation of this strategy is its restriction to presenting only a single antigenic epitope. DNA vaccines cannot amplify and spread in vivo as replicating viral vaccines do which limits their potency. With this in mind, Hung and colleagues created a new strategy that consists in fusing viral protein 22 (VP22) to the HPV-16 E7 proteins. VP22 is a herpes simplex virus-1 (HSV-1) protein that is involved in intracellular and intercellular transport and distributes proteins to many cell types. In this study the vaccine was able to increase MHC-I presentation of antigen through intracellular spreading, leading to increased E7-specific CD8+ T cells and protection against E7 expressing tumor [Hung et al., 2001a]. However, a more recent publication indicated that this increase in immune response was not the result of intracellular spreading [Perkins et al., 2005].
Strategies increasing both MHC-I and MHC-II loading
In a study by Kim and colleagues in 2004, DNA vaccines expressing different ER chaperone proteins linked to antigen were tested for their ability to enhance antigen processing and presentation to T cells in mice [Kim et al., 2004]. Intriguingly, the most potent DNA vaccine was the one encoding calreticulin (Crt) fused to the antigen. This vaccine induced the highest number of tumor-specific CD8+ T cells and potent long-term protection, notably against tumors with low levels of MHC-I expression [Kim et al., 2004]. The mechanism of such efficacy is not well-known. Four years later, the same authors further improved the capacity of this vaccine to induce an immune response against cancer. They prepared two DNA vaccines: the first expressing the Crt linked to HPV-16 E7 proteins (Crt-E7) and the second expressing the Ii, in which the MHC class II-interacting peptide (CLIP) was substituted for PADRE. The co-administration of these two vaccine-induced PADRE specific CD4+ T-cell responses, which again lead to increased levels of HPV 16 E7-specific CD8+ T cells compared with Crt-E7 alone [Kim et al., 2008]. This data proved once again the ability of CD4+ T cells to improve the CD8+ T-cell immunogenicity of DNA vaccines, even when the CD4+ and the CD8+ T-cells are not specific for the same antigen. Thus, both CD4+ and CD8+ T-cell responses are needed to induce an optimal protection against cancer although the CD4+ T cells in this case do not need to be tumor specific. A similar conclusion was reached recently where the provision of the PADRE epitope, together with ER retention, was shown to stimulate potent CD8+ T-cell responses [Oosterhuis et al., 2012]. The Crt-E7 DNA vaccine was further tested using Sindbid virus replicon particles. These vectors may be a promising strategy because of their high level of RNA replication, their capacity to stimulate the innate immune system and the ability to infect a variety of diverse cell types. In addition, they are self-replicating in cells and they are not associated with DNA integration into the host genome. They do however, share the relatively inefficient tissue transduction with DNA vaccines [Cheng et al., 2006].This new approach of Sindbid virus replion particles showed to increase the E7-specific CD8+ T-cell response and generated long-term tumor-specific immunity [Cheng et al., 2006].
As described in the introduction, attempts to improve CD4+ and CD8 + T-cell responses have also been made with replication deficient adenovirus vector encoding the Ii fused to a TAA. The Ii was originally chosen to improve CD4+ T helper cell activation following adenoviral vaccination as Diebold et al. showed that Ii increases MHC-II presentation of the linked antigen [Diebold et al., 2001]. However, this antigen engineering results in an increase of both MHC-I and II antigen presentation on the surface of transduced cells and enhances both CD4+ and CD8+ T-cell responses. The molecular mechanism which leads to increased levels of MHC-I/antigen complex presentation on the surface of transduced cells, i.e. not cross-presentation, has not yet been resolved but it is a subject of current study in our laboratory. This strategy has also the capacity to induce a fast and prolonged immune response similar to those observed following vaccination with live virus and delays the tumor growth in the murine B16.F10 melanoma model [Holst et al., 2008]. In combination with systemically acting monoclonal antibody blockade of CTLA4, the Ii linked vaccine can induce regression of established B16F10-GP melanomas [Sorensen et al., 2010].
Additional mechanisms to induce antitumor immunity by vectored gene fusion
In addition to increasing antigen presentation, several studies using fusion of antigen to cytokines, chemokines or viral proteins targeting cell-surface receptors have shown to overcome the anergy that exists in the tumor [Biragyn et al., 1999, Zhang et al., 2003, Seo et al., 2009, Diniz et al., 2010].
In 1999, Biragyn and colleagues described an interesting study testing a naked DNA vaccine that encodes a self-tumor antigen fused to chemokines (MCP-3 and IP-10). In this study, they showed that the fusion can convert a non immunogenic self tumor antigen to a potent immunogen. In addition, vaccination in mice generated superior protection against a large tumor challenge, as compared with the best available protein vaccines. This was correlated with a high level of anti-self tumor antigen antibody. The mechanism suggested was that the chemokine targets APCs for efficient receptor-mediated uptake and processing of self-tumor antigen. In addition, protection was not induced by the controls such as fusion with truncated chemokines that lack receptor binding [Biragyn et al., 1999].
Another interesting study is the one described by Zhang and colleagues. where they tested an adenoviral vector encoding a TAA (HPV 16 E7 protein) fused to CD40 ligand (CD40L). This approach induced activation of APC T-cell mediated tumor immunity for up to 1 year after vaccination and tumor regression of established TC-1 tumors in C57BL/6. This study also showed that the vaccine encoding a self-tumor antigen fused to CD40L suppresses tumor growth in all vaccinated mice (mice were transgenic for the self-tumor antigen) [Zhang et al., 2003]. Thus, using this strategy, it is possible to induce a long-lasting cellular immune response against self-antigens in anergic animals. Recently, the tumor efficacy of this vaccine has been improved by combining codon optimization of the antigen (likely leading to increased antigen expression) with secretion of the antigen using a signal peptide (likely leading to increased MHC-II presentation and possibly cross-presentation) and electroporation (leading to increased antigen load and local inflammation) [Chiarella et al., 2008]. Each of these additions acted synergistically to improve CD8+ T-cell responses and antitumor immunity [Seo et al., 2009].
The last interesting approach worth mentioning is the therapeutic efficacy of vector vaccines encoding the HSV glycoprotein D (gD) fused to a tumor antigen (HPV 16 E5, E6 and E7 proteins) against HPV-16 associated tumor [Diniz et al., 2010]. The gD is a glycoprotein from the HSV that binds to HSV entry mediator (HVEM) and competes for the same binding site as the B- and T-lymphocyte attenuator (BTLA), which provides an inhibitory signal to B-cells and T-cells [Compaan et al., 2005]. The interaction between gD and HVEM increases DC survival through NFκB activation by inhibiting the binding of HVEM to BTLA [Cheung et al., 2009], which in turn enhances the immune response [Lee et al., 2002, Chen et al., 2006]. All these characteristics make the gD an interesting tool when aiming to increase the immune response. Thus, the authors tested the fusion of the gD to three HPV-16 oncogenic proteins using both naked DNA and adenoviral vector vaccines. Both vaccines induced an increase of tumor antigen specific CD8+ T-cell activation, which confers slight protection against TC-1 cells after 3 doses injected intramuscularly (i.m.) (only tested with DNA vaccine) [Diniz et al., 2010] . Although this strategy shows promising results against cancer, it may not be easily transferable to the clinical setting. Unfortunately, human neutralizing antibodies induced by HSV infection target the same region of the gD that is required for vaccine efficacy [Whitbeck et al., 1999, Chentoufi et al., 2008].
Clinical trials
Pre-clinical studies have provided encouraging evidence of enhanced immune responses and tumor protection against cancer cell lines by DNA fusion gene vaccines, yet only a few strategies employing fusion gene motifs have been tested in clinical trials. Chudley et al. designed a DNA fusion gene vaccine encoding a strongly immunogenic helper domain (DOM) derived from fragment C (FrC) of tetanus toxin and linked to an HLA-A2 binding epitope from prostate-specific membrane antigen (PSMA27). In pre-clinical models, this approach showed to induce durable tumor-specific CD8+ T-cell responses able to kill tumor expressing endogenous PSMA [Vittes et al., 2011]. This strategy is undergoing a phase I/II dose escalation trial in patients with prostate cancer. The results so far show that the vaccine is safe, and generates anti-PSMA specific responses in the majority of patients. The vaccine is delivered using i.m. DNA injection and electroporation (EP) [Chudley et al., 2012].
Trimble and colleagues designed another DNA vaccine encoding signal peptide HPV-16 E7 protein (detox) fused to the Mycobacterium tuberculosis heat shock protein 70. A phase I study investigated this approach administered by i.m. injection at escalating doses. The study showed that the vaccine was well tolerated, however, it failed to induce significant antibody or T-cell responses [Trimble et al., 2009]. The vaccine is now being tested in a heterologous prime-boost strategy (DNA vaccine and viral-vector), and initiatives have been taken to start a clinical trial using the Crt sequence as a genetic adjuvant (Wu TC at the International Conference on papillomaviruses, Puerto Rico 2012).
In summary, advancement in fusion genes and route of delivery has greatly enhanced the immunogenicity of DNA vaccines in pre-clinical models. However, in clinical trials such strategies seem to be less efficient at least when provided as a DNA vaccine. DNA fusion vaccines delivered via electroporation are now under development against PSMA [Chudley et al., 2012], whereas the first clinical test using a viral-vectored fusion-gene vaccine is expected to be used as a partner in a heterologous prime-boost regimen directed against cervical cancer. Strategies that directly combine tumor antigen-specific CD4+ and CD8+ T cells, such as the MHC class II associated invariant chain coupling or the HSV gD fusion, have not yet been clinically tested.
Conclusion
A major challenge has been to develop approaches that induce relevant T cells in patients with established malignant diseases. Often, the cancer has been present for several years when the tumor becomes clinically apparent and the interplay between tumor and immune system in this period have led to poor immune responsiveness against the tumor. Recent advances in our understanding of antigen presentation and tolerance have led to promising strategies, notably using vectored fusion gene vaccines. The aim of this gene-fusion strategy is to enhance the adaptive immune response against the tumor, most especially via increased antigen presentation. Such approaches have distinct advantages in the ability to prime the T cells of low avidity to the TAA that are available in tumor bearing hosts. Preliminary experiments indicate that some of these improvements are also transferable to viral vectored vaccines and the most impressive pre-clinical data have been obtained using viral vectored vaccines and DNA with electroporation. The field now awaits the first clinical efficacy data of a DNA fusion vaccine delivered via electroporation and the first trial of a vaccine expressing TAA fused to a genetic adjuvant boosted by a viral vector.
Footnotes
Acknowledgements
The authors would like to acknowledge the assistance of Delphine Ragonnaud for the illustrations as well as Thomas Mandel Clausen, Liz Stevenson and Anne-Marie Andersson for their comments to the manuscript.