
Abstract
The overall objective of these guidelines is to provide evidence-based recommendations for the diagnosis and management of immunoglobulin G4 (IgG4)-related digestive disease in adults and children. IgG4-related digestive disease can be diagnosed only with a comprehensive work-up that includes histology, organ morphology at imaging, serology, search for other organ involvement, and response to glucocorticoid treatment. Indications for treatment are symptomatic patients with obstructive jaundice, abdominal pain, posterior pancreatic pain, and involvement of extra-pancreatic digestive organs, including IgG4-related cholangitis. Treatment with glucocorticoids should be weight-based and initiated at a dose of 0.6–0.8 mg/kg body weight/day orally (typical starting dose 30-40 mg/day prednisone equivalent) for 1 month to induce remission and then be tapered within two additional months. Response to initial treatment should be assessed at week 2–4 with clinical, biochemical and morphological markers. Maintenance treatment with glucocorticoids should be considered in multi-organ disease or history of relapse. If there is no change in disease activity and burden within 3 months, the diagnosis should be reconsidered. If the disease relapsed during the 3 months of treatment, immunosuppressive drugs should be added.
Keywords
Introduction and methodology
Aim of the guidelines
The overall objective of these guidelines is to provide evidence-based recommendations for the diagnosis and management of IgG4-related digestive disease in adults and children. Target users of the guidelines are clinicians involved in the care of patients with IgG4-related digestive disease.
Literature review
A comprehensive literature search for relevant articles was performed using the PubMed, Embase, and Cochrane databases was conducted.
The following keywords were used in various combinations: “pancreas” OR “pancreatic” OR “pancreatitis” AND “autoimmune” OR “IgG4” OR “rheumatoid”; “cholangitis” OR cholagiopathy”; “other organ involvement” OR “systemic disease”. Furthermore, additional keywords were used by the working parties to accommodate their specific topics, e.g. “therapy”, “treatment”, children”, “pediatric”, “kidney”, etc. The search was limited to human subjects with language restriction to English studies until 1 September 2019. The snowball strategy, including a manual search of the references listed by studies retrieved from the online databases and from previously published reviews, was also performed to identify potential additional studies. The following inclusion criteria were used: (a) randomized or observational cohort studies, including systematic reviews, on patients with IgG4-related digestive disease, which focused on specific study questions; (b) studies published in the English language; and (c) studies available in full text.
In view of the relatively small number of studies on IgG4-related digestive disease, which is rare in everyday clinical practice, even non-randomized studies with less than 20 patients were used.
Recommendations, grades of evidence and outcome reporting
The recommendations format comprised the question, the statement, its level of evidence, strength of recommendation, and the percentage agreement of the global consensus group with the final version. Statements are followed by qualifying comments, written by each working party (WP) (a list of abbreviations is part of the supplement) and reviewed by the entire scientific board (executive committee). Relevant comments and suggestions made by the global consensus group (expert readers) were also taken into consideration.
The Grading of Recommendations Assessment, Development, and Evaluation (GRADE) system was applied (Table S1). All participants and reviewers used a GRADE system tutorial (link on UpToDate: http://www.uptodate.com/home/grading-tutorial).
The final outcomes of the systematic reviews were discussed among the members of each WP.
The review groups provided the following for each clinical question:
Recommendation: the GRADE strength of recommendation (1=strong, 2=weak) and the quality of evidence (A=high, B=moderate, C=low).
The first meeting of the group had taken place during the United European Gastroenterology Week (UEGW) in Vienna, Austria (October 2018). The working groups were finalized and a responsible leader for each group was named together with a time manager. The Conflict of Interest forms were distributed to all participants and signed scanned copies were sent to United European Gastroenterology (UEG) central in Vienna according to UEG rules. After the meeting in Vienna, we determined WPs, members of the WP (Table S2) and proposals for questions. After all WPs completed the first draft of the guidelines, the questions and statements were distributed among the entire expert group. The questions and answers, including related comments, were uploaded to the Delphi platform and voted upon. All questions with less than 80% agreement were discussed at a meeting during the European Pancreatic Club meeting in Bergen, Norway (June 2019) with Test and Evaluation Directorate (TED) voting. The comments to all questions, and particularly those with less than 80% agreement during the TED voting, were returned to the WP. A second round of the Delphi voting was performed during autumn 2019 and the final round of discussion, including TED voting, was performed during the UEG week in Barcelona, Spain (October 2019). Following the consensus reached after the UEG week (2019) and a final round of adjustments, a first draft of the manuscript was prepared in December 2019 and was sent out to external readers and finalized according to the comments received. In addition to this written version, an interactive smartphone app was developed (free download).
The working group received endorsements and funding from UEG with Swedish Society of Gastroenterology (SGF) as the National Society leading the development of these guidelines.
Overview
Biomarkers in IgG4-related gastrointestinal diseases IgG4-related disease of pancreas IgG4-related diseases of liver and bile ducts IgG4-related gastrointestinal diseases of esophagus, stomach, and bowel Clinical manifestations and management of systemic IgG4-related diseases IgG4-related digestive diseases in children IgG4-related gastrointestinal diseases and diabetes mellitus IgG4-related gastrointestinal diseases and cancer Systemic treatment of IgG4-related digestive diseases
WP 1: Biomarkers in IgG4-related gastrointestinal disease
Q1.1 Are there serum biomarkers that can be measured to establish the diagnosis of IgG4-related gastrointestinal disease?
Statement 1.1: IgG4 serum level alone lacks sensitivity and specificity, but can be helpful to establish the diagnosis, and therefore should be measured if IgG4-related gastrointestinal disease is suspected. (GRADE 2C; strong agreement)
Q1.2. Does the measurement of IgG4 help to monitor the disease course?
Statement 1.2: The measurement of IgG4 serum levels to monitor the disease course may be helpful in some patients. (GRADE 2C, strong agreement)
Q1.3. Is the measurement of the serum carbohydrate antigen (CA) 19-9 level recommended to differentiate autoimmune pancreatitis (AIP) from pancreatic adenocarcinoma?
Statement 1.3: Elevated serum CA 19-9 levels are influenced by factors, such as cholangitis, and when used alone CA 19-9 displays limited accuracy in differentiating AIP from pancreatic adenocarcinoma, but given that it is an easy-to-perform and cheap test with acceptable sensitivity and specificity, its use integrated with other second-level diagnostics (e.g. biopsy, computed tomography (CT) scan) is encouraged. (GRADE 2C; strong agreement)
(Statement 1.3): Value of CA 19-9 to differentiate AIP from pancreatic cancer.
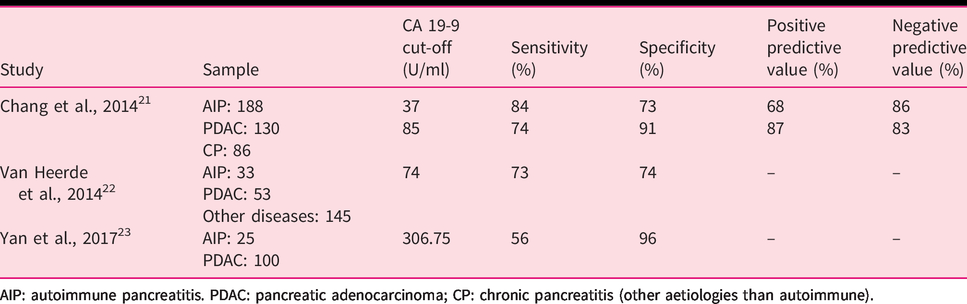
AIP: autoimmune pancreatitis. PDAC: pancreatic adenocarcinoma; CP: chronic pancreatitis (other aetiologies than autoimmune).
WP 2: IgG4-related disease of the pancreas
Q2.1: What are the pathological characteristics of AIP type 1?
Statement 2.1: There are four key histological features of AIP type 1: (a) lymphoplasmacytic infiltration affecting the tissue either diffusely or in a patchy manner, (b) storiform fibrosis, (c) obliterative phlebitis, and (d) increased numbers of IgG4+ plasma cells. (GRADE 1B; strong agreement)
While AIP type 2 shares several features with AIP type 1 (see Table 2), the presence of few or no IgG4+ plasma cells in combination with the presence of granulocytic epithelial lesions (GELs) is considered confident histological evidence. GELs are characterized by the infiltration of neutrophilic granulocytes in the duct epithelial lining, causing degenerative epithelial changes, often including epithelial detachment. The presence of an acinar infiltrate, (including neutrophils), in the absence of GELs or an elevated IgG4+ plasma cell count (≤10/HPF) is regarded as a probable diagnosis of AIP type 2. 1
(Statement 2.1): Diagnostic microscopic features of type 1 and type 2 autoimmune pancreatitis (adapted from Zhang et al. 226 ).

AIP: autoimmune pancreatitis; GEL: granulocytic epithelial lesion
Q2.2: What are the imaging features of AIP?
Statement 2.2: The classical imaging features of AIP are parenchymal enlargement, ‘sausage-like’ shape, peripancreatic edematous rim, and main pancreatic duct narrowing without upstream dilatation. These features may be diffuse or focal but can also be highly variable. (GRADE 2C; strong agreement)
Parenchymal changes suggestive of AIP are:
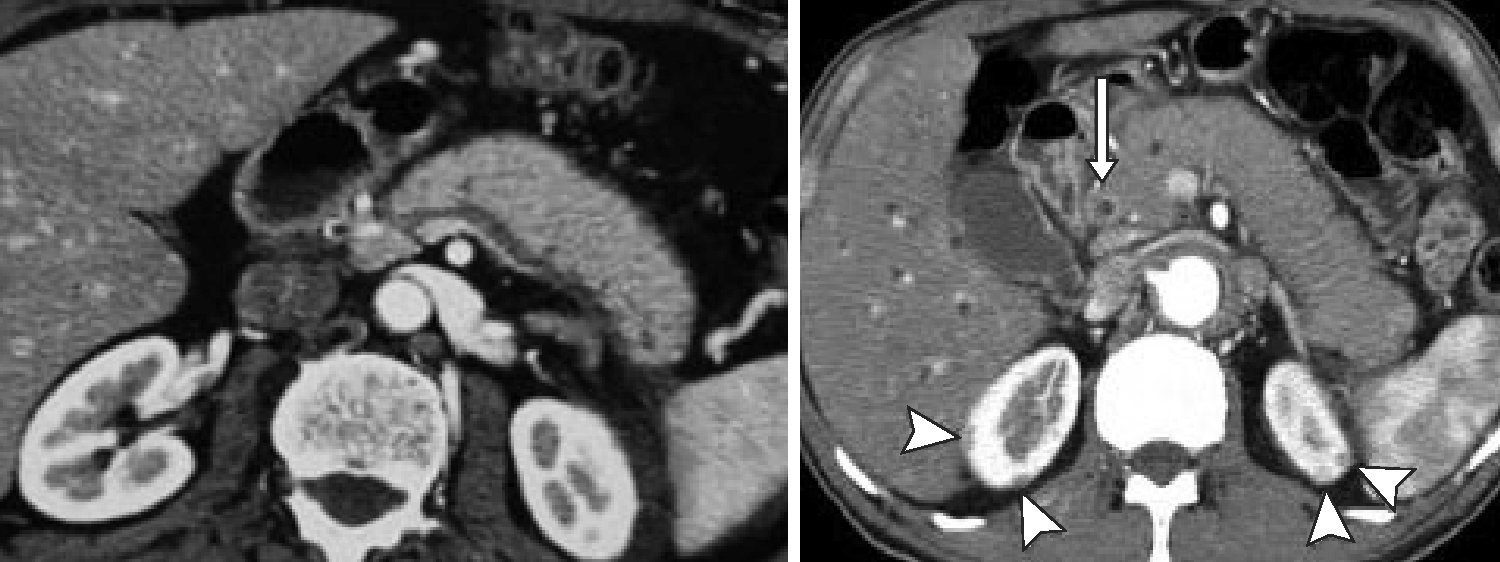
Diffuse or (multi-) focal enlargement with loss of the normal multilobulated pattern (‘sausage-like’ shape); with diffuse involvement, more frequent in type 1 and focal involvement in AIP type 2 (Figure 1).
CT pictures of IgG4-RD in the abdomen. Sausage-like pattern of the pancreatic gland with loss of lobulation (left). Contrast enhancement of the distal bile duct (CBD, arrow) indicating IgG4-related cholangitis (IRC). Note the typical kidney lesions (arrowhead) pathognomonic to IgG4-RD underscoring the diagnosis.
225
Altered imaging characteristics, such as lower signal intensity (SI)/echogenicity on unenhanced T1-w MRI/(E)US, respectively, moderately higher SI on T2-w MRI, impeded diffusion on MRI, and increased 18F-fluorodeoxyglucose (FDG)-uptake on PET-CT compared with normal parenchyma. Post injection of (iodine-, gadolinium-, or microbubble-based) contrast media, there is dotted/patchy enhancement in the late arterial/pancreatic phase that progressively increases towards the later vascular phases. Rectangular shape of the tail (‘cut-tail sign’). Thin peripancreatic edematous rim or progressively enhancing true capsule.
Ductal changes suggestive of AIP are:
Long-segment (i.e. ≥1/3 of the length) or multifocal main pancreatic duct (MPD) involvement (narrowing or vanishing) without upstream dilatation or other signs of obstructive pancreatitis. Skip lesions, i.e. ≥2 involved MPD-segments separated by a normal MPD-segment. ‘Duct-penetrating’ (i.e. visible MPD- and/or common bile duct (CBD)-lumen) and ‘icicle’ (i.e. a progressive decrease of MPD-diameter) signs within an enlarged parenchymal area.
Q2.3: What is the role of endoscopy in diagnoses of AIP type 1?
Statement 2.3: Endoscopic ultrasound (EUS) provides pancreatic imaging findings suggestive of AIP and is used for obtaining tissue samples for the histological diagnosis of the disease. (GRADE 2B; strong agreement)
AIP must be differentiated from pancreatic carcinoma.1,42–46 Pancreatography findings, such as a long narrowing of the main pancreatic duct (>1/3 the length of the MPD), lack of upstream dilatation, skipped narrowed lesions, and side branches arising from the narrowed portion, suggest AIP rather than pancreatic carcinoma.1,42–45 IgG4-immunostaining of biopsy specimens obtained from the major duodenal papilla supports the diagnosis of AIP.1,43,45 IRC must be differentiated from cholangiocarcinoma and primary sclerosing cholangitis (PSC).43–45 Specific cholangiography findings support the diagnosis of IRC (see below).43–45 IgG4-immunostaining of trans-papillary biopsy specimens of bile duct strictures may help exclude cholangiocarcinoma and support the diagnosis of IRC.43–45 EUS may demonstrate diffuse hypoechoic pancreatic enlargement and other features of pancreatic disease in patients with AIP.43–45 EUS-guided tissue acquisition is used for obtaining adequate tissue samples for the histological diagnosis of AIP and to exclude pancreatic carcinoma.1,43–45 EUS-guided tissue acquisition with a core biopsy, with a 19-gauge needle, is recommended, but even use of a 22-gauge needle for a sample allowing histological evaluation can be obtained.43–45 EUS and intraductal ultrasonography may show wall thickening of the CBD in patients with IRC (see below).43–45
Q2.4: What is the role of surgery in AIP type 1?
Statement 2.4: Surgery is generally not indicated for AIP type 1. Surgery might be considered in patients when suspicion of pancreatic cancer cannot be excluded after complete diagnostic work-up. (GRADE 2B; strong agreement)
Q2.5: What is the expected outcome and optimal follow-up of patients with AIP type 1?
Statement 2.5.1: AIP is a special and treatable form of chronic pancreatitis with a good response to initial glucocorticoid therapy, but high rates of disease relapses. Other organ involvement (OOI), defined as the presence of extra-pancreatic disease, is common. (GRADE 1A; strong agreement)
Statement 2.5.2: Long-term sequelae, such as exocrine and endocrine insufficiency, often occur in patients with AIP type 1. (GRADE 1B; strong agreement)
Statement 2.5.3: Screening for a deficiency of fat-soluble vitamins (A, D, E, and K), zinc, calcium, and magnesium should be considered in line with UEG evidence-based guidelines for the diagnosis and therapy of chronic pancreatitis (HaPanEU). 54 (GRADE 2A; strong agreement)
Statement 2.5.4: Life-long follow-up of patients with AIP type 1 is advisable. (GRADE 2C; strong agreement)
WP 3: IgG4-related diseases of liver and bile ducts
Q3.1. What is the definition and proposed nomenclature of IgG4-related hepatobiliary disease?
Statement 3.1: The most common manifestation of IgG4-related hepatobiliary disease is IgG4-related cholangitis. (GRADE 2C; strong agreement)
Q3.2. What are the clinical, biochemical, pathological, and radiological characteristics leading to the diagnosis of IgG4-related cholangitis?
Statement 3.2: Jaundice, a cholestatic serum enzyme profile, includes elevated serum IgG4 concentrations, histological features (including lymphoplasmacellular infiltrates with >10 IgG4+ plasma cells/HPF, storiform fibrosis, and/or obliterative phlebitis), and extrahepatic, hilar, and/or intrahepatic bile duct strictures, which are characteristic features of IgG4-related cholangitis. (GRADE 2C; strong agreement)
The diagnosis of IRC may be difficult without histological sampling and, as it happens under ongoing steroid therapy, since then IgG4 may be already within normal range.80,81 The measurement of IgG subclass 2 (IgG2) may help in confirming the diagnosis of IRC, as demonstrated in a recent study. 82
Cholangiographic characteristics and a classification are shown in Figure 2. 83 Lower strictures of the CBD without strictures of upstream bile ducts represent the most common finding (type 1). Intrahepatic segmental (type 2a) and diffuse (type 2b) strictures, in addition to a lower CBD stricture, when taken together represent the second most common finding. The combination of hilar and lower CBD strictures (type 3) and hilar strictures only (type 4) are additional variants. 83
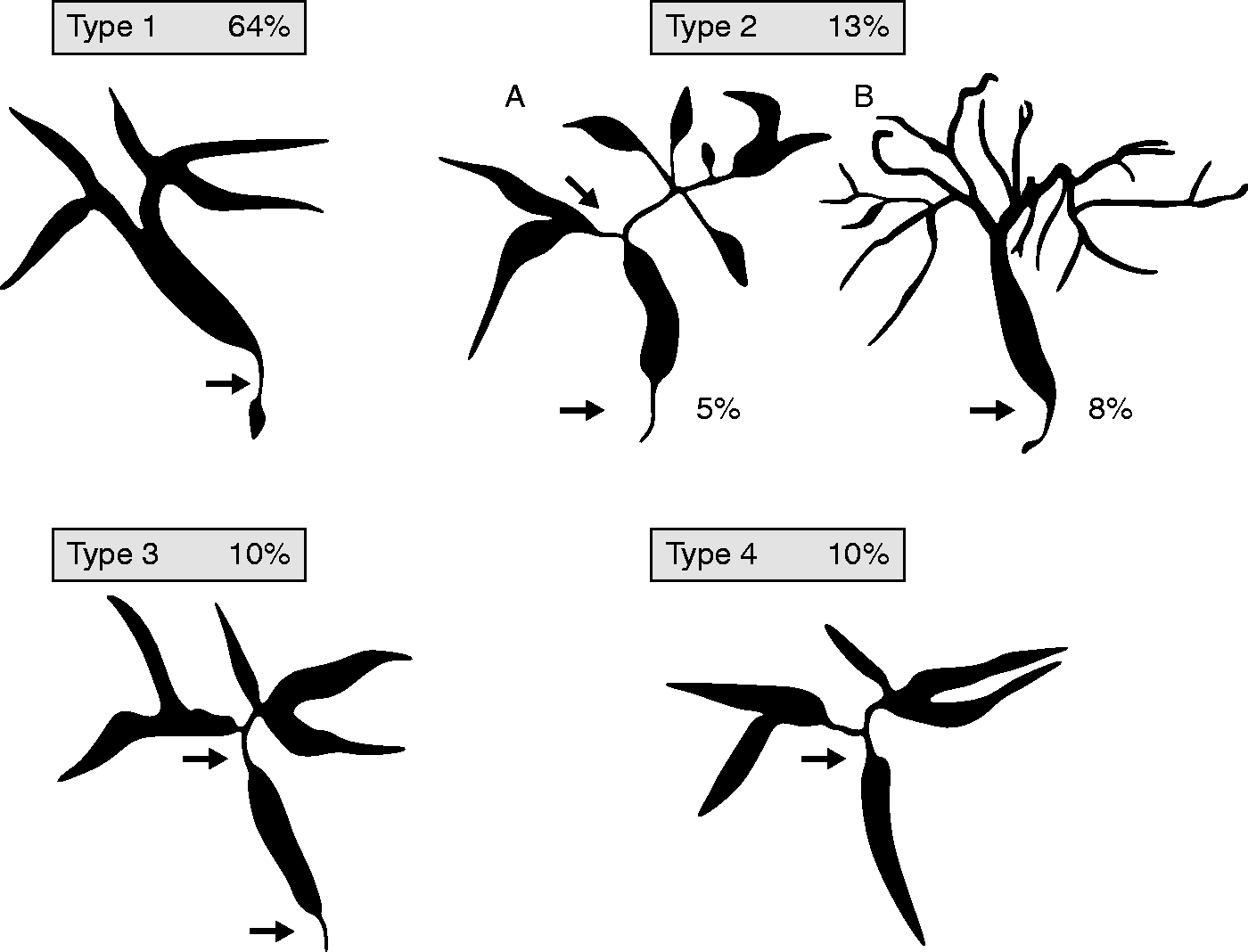
Classification of IgG4-related cholangitis 83 (related to statement 3.2).
Q3.3. Is glucocorticoid treatment response indispensable for the diagnosis of IgG4-related hepatobiliary disease?
Statement 3.3: Treatment response is regarded as a major diagnostic criterion but is not indispensable for the diagnosis of IgG4-related cholangitis. (GRADE 2C; strong agreement)
Q3.4. What are the evidence-based manifestations of IgG4-related hepatobiliary disease in addition to IgG4-related cholangitis?
Statement 3.4: IgG4-related cholangitis and IgG4-related hepatic pseudotumours are hepatic manifestations within the spectrum of IgG4-related disease. Other histopathological features of liver tissue might also be interpreted as reactive changes due to IgG4-related cholangitis or autoimmune pancreatitis. (GRADE 2C; strong agreement)
WP 4: IgG4-related gastrointestinal disease of esophagus, stomach, and bowel
Q4.1. How often do IgG4-related diseases occur in the esophagus, stomach, and bowel?
Statement 4.1: Involvement of esophagus, stomach, and bowel in IgG4-related disease is rare or non-existing. (GRADE 2C; strong agreement)
Q4.2. What are the typical clinical features and diagnostic criteria of IgG4-related disease in the esophagus, stomach, and bowel?
Statement 4.2.1: Typical clinical features and diagnostic criteria of IgG4-related disease of the esophagus, stomach, and bowel have only rarely been reported – and the reported cases are often incomplete regarding diagnostic criteria. (GRADE 2C; strong agreement)
Q4.3: What should be the treatment approach for IgG4-related disease of the esophagus, stomach, or bowel?
Statement 4.3.1. Pharmacological therapy of IgG4-related disease of the gut is based on the same principles as IgG4-related disease of other organs. (GRADE 2C; strong agreement)
Statement 4.3.2. In a patient with gastrointestinal mass lesion and equivocal/nondiagnostic histology for IgG4-related disease with negative malignant cells, empirical treatment with glucocorticoid for 1 month may be a suitable option. (GRADE 2C; strong agreement)
WP 5: Clinical manifestations and management of systemic IgG4-related diseases
Q5.1 What is the spectrum of organ involvement and clinical presentations in IgG4-related disease?
Statement 5.1.1: Clinical manifestations of IgG4-related disease are extremely variable depending on the type and number of organ/tissues involved. (GRADE 1A; strong agreement)
Statement 5.1.2: IgG4-related disease is a systemic condition typically involving two or more organs. (GRADE 1B; strong agreement)
Statement 5.1.3: The most frequently involved organs are: the pancreato-hepatobiliary tract, salivary and lacrimal glands, the retroperitoneum, kidneys, lungs, and aorta. (GRADE 1A; strong agreement)
Q5.2 What is the optimal diagnostic work-up and follow-up strategy for IgG4-related disease?
Statement 5.2.1: The most accurate diagnostic assessment of IgG4-related disease is based on a full clinical history, physical examination, laboratory investigations, pathology, and imaging studies. Life-long follow-up of patients with IgG4-related disease is advisable. (GRADE 1B; strong agreement)
Statement 5.2.2: Whenever possible the diagnosis of IgG4-related disease should be confirmed by pathological examination from a guided biopsy. (GRADE 1A; strong agreement)
Statement 5.2.3: Patients with systemic manifestations of IgG4-related disease should be followed over time with specific serological exams and imaging studies depending on the spectrum of organ involvement, as well as with the IgG4-RD Responder Index. (GRADE 1C; strong agreement)
Q5.3 How do we assess disease activity and differentiate chronic damage from active lesions in IgG4-related disease?
Statement 5.3.1: There is no reliable biological marker to assess disease activity on its own. (GRADE 1A; strong agreement)
Statement 5.3.2: IgG4-RD Responder Index can assess changes in multi-organ disease activity and is now being used in multicentre clinical trials. (GRADE 1C; strong agreement)
Statement 5.3.3: The most accurate assessment of IgG4-related disease activity relies on the combination of findings from physical examination, laboratory exams, histology, and imaging studies. (GRADE 1B; strong agreement)
WP 6: IgG4-related digestive diseases in children
Q6.1: What is the prevalence of IgG4-related digestive disease in children?
Statement 6.1: There are currently insufficient data regarding prevalence of IgG4-related digestive disease in children. IgG4-related digestive disease is extremely rare in childhood. The most common IgG4-related digestive disease in the paediatric population is AIP type I, which is rare, but the accurate prevalence remains unknown. (GRADE 2C; strong agreement)
Q6.2: What is the difference in diagnosis of IgG4-related digestive diseases in childhood compared with adults?
Statement 6.2.1: There are currently insufficient data regarding differences in diagnosis of IgG4-related digestive disease in children. The diagnosis of IgG4-related digestive disease in children should be based on adult criteria, in the absence of paediatric consensus on diagnostic criteria. (GRADE 2C; strong agreement)
Q6.3: What are the differences in approaches to treatment of IgG4-related digestive disease in children as opposed to adults?
Statement 6.3.1: There are currently insufficient data regarding different treatments of IgG4-related digestive diseases in children. (GRADE 2C; strong agreement)
Statement 6.3.2: There are currently insufficient data on the differences in treatment of IgG4-related liver disorders in children compared with adults. (GRADE 2C; strong agreement)
Indications for ERCP with balloon dilatation are limited and may be considered individually in cases of dominant or symptomatic biliary strictures.164,165
Q6.4: What are the clinical manifestations of AIP type I in children?
Statement 6.4.1: The classic form of AIP (type 1) is rarely diagnosed in childhood. The diagnosis of AIP, in the absence of paediatric consensus on diagnostic criteria, should be carried out at a specialized paediatric pancreatic centre based on adult criteria. There is currently insufficient data about transition from AIP to chronic pancreatitis in the paediatric population. (GRADE 2C; strong agreement)
Statement 6.4.2: Children with AIP type 1 may present acutely with jaundice, pancreatic mass, pain, vomiting, and weight loss. Patients usually respond well to glucocorticoid therapy with a lower likelihood of recurrence. Some paediatric patients may exhibit resolution of symptoms without any treatment. (GRADE 2C; strong agreement)
Statement 6.4.3: Transabdominal ultrasonography is recommended as the ‘first step’ in a diagnostic work-up. In the suspicion of AIP, presence of pancreas enlargement, or pancreatic mass lesions with jaundice, magnetic resonance cholangiopancreatography (MRCP) and/or EUS is recommended. (GRADE 2C; strong agreement)
Statement 6.4.4: Pancreatic biopsy is not necessary to start immunosuppressive treatment. (GRADE 2C; strong agreement)
Statement 6.4.5: Glucocorticoids are the first line of treatment for remission induction, unless there are contraindications for their use. Children with AIP type 1 inflammation with low disease activity at the beginning of treatment do not require any maintenance treatment. (GRADE 2C; strong agreement)
INSPPIRE recommends oral prednisolone as a first-line treatment at a dose of 1–1.5 mg/kg/day to maximum 40–60 mg given in one or two divided doses for 2–4 weeks. Thereafter, the dose should be tapered. In case of relapse, a new course of prednisolone is recommended. 166
WP 7: IgG4-related digestive disease and diabetes mellitus
Q7.1 What is the prevalence of diabetes mellitus in IgG4-related disease of the pancreas?
Statement 7.1: Diabetes mellitus is very common in IgG4-related disease of the pancreas (AIP type 1). Its prevalence ranges between 21% and 77%. (GRADE 2C; strong agreement)
Q7.2 Are there any features of IgG4-related disease of the pancreas associated with the risk and/or severity of diabetes mellitus?
Statement 7.2: Among patients with IgG4-RD of the pancreas (AIP type 1), radiologically defined pancreatic atrophy, pancreatic exocrine insufficiency, age, and smoking are all associated with a significantly higher risk of diabetes mellitus, while there are no specific data on features associated with the severity of diabetes mellitus. (GRADE 2C; strong agreement)
Q7.3. What is the impact of glucocorticoid/immunosuppressive treatment on the risk and/or severity of diabetes mellitus in IgG4-related disease of the pancreas?
Statement 7.3: Glucocorticoid treatment of IgG4-related disease of the pancreas (AIP type 1) induces beneficial effects on the clinical course of diabetes mellitus in approximately 50–60% of all cases. Patients with simultaneous-onset diabetes mellitus show greater glucocorticoid responsiveness than patients with pre-existing diabetes mellitus. (GRADE 2C; strong agreement)
WP 8: IgG4-related digestive disease and cancer
Q8.1 What is the risk of cancer development in the context of IgG4-related disease?
Statement 8.1: IgG4-related disease, and particularly AIP, may be associated with an increased risk of developing malignant disease compared with the general population. Life-long surveillance in patients with IgG4-related disease is advised. (GRADE 2C; strong agreement)
(Statement 8.1): Prevalence rates of malignant diseases in patient cohorts with IgG4-related disease or in patient cohorts with AIP.
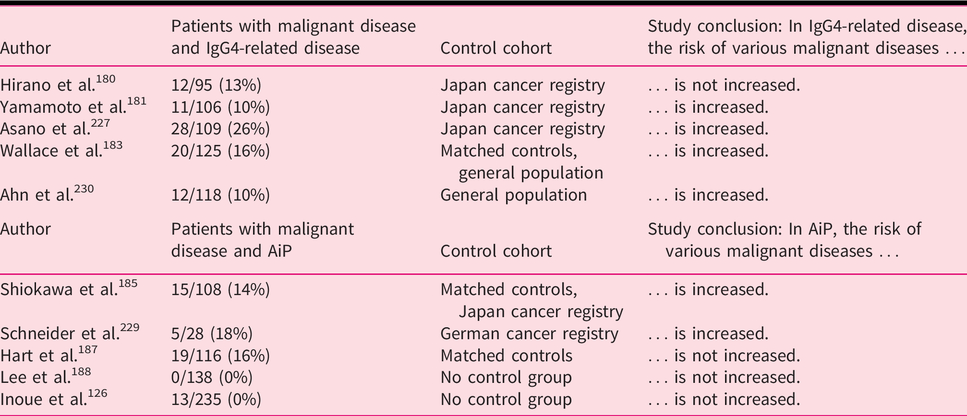
Q8.2. How can we distinguish IgG4-related disease and cancer clinically and radiologically?
Statement 8.2.1: There are no specific clinical features or blood tests that can differentiate between IgG4-related disease and cancer. (GRADE 2B; strong agreement)
Statement 8.2.2. Radiological differentiation between IgG4-related disease and cancer is challenging. A few imaging findings may aid in the differential diagnosis. (GRADE 2C; strong agreement)
The most challenging radiological diagnosis is differentiating between focal AIP and pancreatic cancer, and isolated IgG4-SC (type IV) and extrahepatic cholangiocarcinoma, respectively. The combination of a few MR/CT features may be useful (Table 1).41,190–193 PET/CT may identify a diffuse uptake in the pancreas in focal AIP and show evidence of other organ involvement.41,190 The role of imaging in the differentiation between IgG4-related lymphadenopathy and IgG4-related retroperitoneal fibrosis (RPF) from malignancy (e.g. lymphoma or retroperitoneal malignancies) has been poorly investigated and is still unclear. A biopsy and clonality assessment are often required to clarify the diagnosis and may still be inconclusive.
Q8.3. How can we distinguish IgG4-related disease and cancer histologically?
Consensus Statement on the Pathology of IgG4-RD (histological Boston criteria):
Includes three key morphological features: dense lymphoplasmacytic infiltrate, fibrosis that is at least focally storiform, and obliterative phlebitis. Additional morphological features: phlebitis and increased eosinophils. The number of IgG4-positive plasma cells are measured as the mean IgG4+ plasma cell count per HPF within the three HPFs containing the greatest number of these cells when the distribution is patchy. If the IgG4-positive plasma cells are distributed diffusely, the mean IgG4+ plasma cell count is determined in three random HPFs.
Histologically highly suggestive of IgG4-related disease: Two or more key morphological features and immunohistochemistry demonstrating IgG4-positive plasma cells above 30–100 per HPF (organ-specific cut-off in resection specimen) or 10–200 per HPF (organ-specific cut-off in a biopsy specimen) and IgG4/IgG plasma cell ratio of over 40%.
Probable histological features of IgG4-related disease: One key morphological feature and immunohistochemistry demonstrating IgG4-positive plasma cells above 30–100 per HPF (organ-specific cut-off in resection specimen) or 10–200 per HPF (organ-specific cut-off in a biopsy specimen) and IgG4/IgG plasma cell ratio of over 40%.
Statement 8.3.1: In resection specimens, there are established histological criteria to distinguish cancer and IgG4-related disease (the so-called Boston criteria: three morphological features of lymphoplasmacytic infiltrate, storiform fibrosis, obliterative phlebitis, and in addition, immunohistochemistry demonstrating IgG4-positive plasma cells above 50–100 per HPF (organ-specific cut-off) and IgG4/IgG ratios of over 40%). (GRADE 2B; strong agreement)
Statement 8.3.2. In biopsy specimens, distinguishing IgG4-related disease and cancer is more challenging. Risk of sampling error should be considered in a negative biopsy. Non-specific inflammation, with increased IgG4-positive cells can occur in both cancer and IgG4-related disease. (GRADE 2C; strong agreement)
Statement 8.3.3. An increased frequency of synchronous or metachronous lymphomas and IgG4-related disease has been reported. Immunohistochemical, flow cytometric, and molecular studies may help to differentiate them. (GRADE 2C; strong agreement)
Q8.4. Is IgG4-related disease a paraneoplastic condition?
Statement 8.4.1: Malignancy in patients with IgG4-related disease, and particularly AIP, is most often identified in organs distinct from those affected by the disease. (GRADE 2B; strong agreement)
Statement 8.4.2: IgG4 antibodies can be found in both the context of IgG4-related disease and in several patients with cancer. Further studies are needed to establish the relationship between IgG4 antibodies, IgG4-related disease, and cancer. (GRADE 2C; strong agreement)
Cancer and pancreatic tissues of AIP patients with cancer share key immune responses leading to the enhancement of IgG4 antibody production. 185 IgG4 antibodies may be specifically generated in response to malignant disease and represent a mechanism of tumour-induced immune escape. In human melanoma patients, 203 tumour-associated B cells are stimulated by a Th2 (IL-10 secreting) tumour microenvironment and polarized to produce IgG4. Strikingly, IgG4 antibodies can inhibit the anti-tumour effector functions of IgG1 antibodies, and IgG4 serum levels are associated with decreased patient survival. 203 IgG4 responses have also been reported in other cancers, including extrahepatic cholangiocarcinoma, pancreatic cancer, and glioblastoma, and elevated serum IgG4 levels have been associated with poorer prognosis in malignant melanoma and biliary tract cancers.
WP 9: Systemic treatment of IgG4-related digestive disease
Q9.1 What are indications and modalities of initial systemic treatment of IgG4-related gastrointestinal diseases?
Statement 9.1.1: All symptomatic patients (e.g. suffering from pancreatic pain, obstructive jaundice) should be considered for treatment, sometimes urgently in cases of organ insufficiency. Treatment can also be proposed to asymptomatic patients in case of: (1) persistence of a pancreatic mass in imaging to rule out cancer, (2) persistence of liver test abnormalities (cholestasis) in case of associated IgG4-related cholangitis, and (3) in subclinical situations that could lead to severe or irreversible organ failure. (GRADE 1C)
Statement 9.1.2: There is no relevant data to support that a treatment should be proposed in patients with AIP without symptoms, just to limit the risk of exocrine or endocrine insufficiencies. (GRADE 1C)
Statement 9.1.3: Treatment with glucocorticoids should be initiated in a weight-based manner at a dose of 0.6–0.8 mg/kg body weight/day orally (typical starting dose 30–40 mg/day prednisone equivalent) for 1 month to induce remission. Response to initial treatment should be assessed at week 2–4 with clinical, biochemical, and morphological markers. Glucocorticoid therapy should gradually be tapered by 5 mg every two weeks (tapering duration 3–6 months). (GRADE 1C)
Glucocorticoids remain the most effective initial treatment, although there are limited clinical trials on the effectiveness of glucocorticoid maintenance therapy.14,16 Glucocorticoids are the preferred first-line medication for active IgG4-related disease, with response rates around 97–100% and a significant decrease of serum IgG4 levels. 14 Treatment with glucocorticoids should be initiated weight-based at a dose of 0.6–0.8 mg/kg body weight/day orally for 1 month to induce remission, but initial glucocorticoid dose can be adjusted. This adjustment must be based on body weight, in case of particularly aggressive disease (use initial dosages above 40 mg/d), or in case of elderly patients, with very mild clinical symptoms (use <20 mg/d). In patients with diabetes, it is important to optimize diabetic control and osteoporosis medication prior to initiation of glucocorticoids, if this is possible. A few data on remission rates using low glucocorticoid doses (e.g. equivalent prednisolone dosing of 10–20 mg/day) are available,14,16 but are too preliminary to be proposed. Response to glucocorticoids has become part of the diagnostic criteria. 1 This suggests that biliary stenting is not mandatory in combination with glucocorticoids in cases of obstructive jaundice to limit the risk of cholangitis. 1 Jaundice totally resolved in less than 15 days with glucocorticoids (without stenting) with a rapid normalization of serum liver tests. Biliary stenting may even propagate pancreatic stone formation in IRC and AIP. 207 As shown by Yukutake et al., serum liver test abnormalities are normalized in 80% and 100% at 15 and 21 days, respectively.208–210 Despite the effectiveness of glucocorticoids, about one-third of patients experience disease relapse during glucocorticoid tapering, thus requiring re-induction therapy. 14 This re-induction is generally managed with an increase in glucocorticoid dosing followed by a prolonged taper. Relapse may occur in the same organ being treated or, interestingly, in previously uninvolved organ systems.14,75 Recurrence occurs more often in patients without prior glucocorticoid therapy (about 40%) than in cases after previous glucocorticoid therapy (about 25%). 14 In the Asian region, especially in Japan and China, a 12-month maintenance therapy is therefore recommended. To be weighed against this are potential side effects of long-term glucocorticoid maintenance therapy against the recurrence rate (about 5% with maintenance therapy versus 22% without maintenance therapy). 211 No relevant data are available to recommend maintaining low-dose therapy for a few months. But several experts recommend maintaining glucocorticoids at ≤10 mg/day (equivalent to 2.5–10 mg/day prednisolone) for 12 months. Some Japanese centres continue low-dose (5 mg) prednisolone for as many as 3 years 212 and beyond. 213
Q9.2. What are the indications for immunosuppressant treatments for IgG4-related gastrointestinal disease?
Statement 9.2: Adding immunosuppressive agents should be considered in case of disease relapse as maintenance of remission strategy, and in patients with a high risk of disease relapse, particularly in the case of multi-organ involvement. If there is no change in disease activity or the disease relapsed during the 3 months of treatment (during glucocorticoid taper or discontinuation), then immunosuppressive drugs should be added. (GRADE 2C)
Q9.3. Which immunosuppressant (for which patient) should be proposed as treatment of IgG4-related digestive disease and what are the secondary effects of the systemic treatment?
Statement 9.3.1: Rituximab should be considered if patients are resistant or intolerant to high-dose glucocorticoids to maintain remission or have failed to respond to immunosuppressive therapies. Dosing protocol (375 mg/m2 body surface area) is used weekly for 4 weeks, followed by infusions every 2–3 months or at two 1000 mg infusions 15 days apart every 6 months. (GRADE 2A)
In IgG4-related digestive disease, rituximab proved effective when administered both at 375 mg/m2 weekly for 4 weeks followed by maintenance infusions every 2–3 months (onco-haematological protocol) or at two 1000 mg infusions 15 days apart every 6 months (immunological/rheumatoid arthritis protocol). 216 Maintenance therapy with rituximab, continued for up to 2 years, was associated with longer relapse-free survival. Adverse events, such as infusion reactions, hypogammaglobulinemia, and severe infections must be noted. Rituximab presents several advantages, such as a glucocorticoid-sparing effect, existing data on remission induction, and (possibly) better safety profile than glucocorticoids or immunosuppressants.
Statement 9.3.2: Immunosuppressive drugs. Immunosuppressants used include: thiopurines (azathioprine and 6-mercaptopurine), mycophenolate mofetil, methotrexate, or calcineurin inhibitors (tacrolimus and cyclosporine A). (GRADE 2A)15,217–219
Therapy with mycophenolate mofetil (MMF). A recent Chinese randomized control trial showed that maintenance therapy with mycophenolate, in addition to glucocorticoids, reduced the risk of relapse (21% at 12 months) compared with glucocorticoids alone (40%), with no increase in adverse events. 221 Therapy with MMF should commence with 1 g/day and can be increased to 1.5–2 g/day under close monitoring of complete blood count. Like azathioprine, many patients have been reported to relapse on low doses of MMF (1 g daily).
Therapy with methotrexate (MTX). Several series of cases reported the role of MTX in patients with relapsing IgG4-related disease . 219 In 10 patients, oral or subcutaneous MTX was introduced on average 5 weeks (range 1–16) after initiation of oral glucocorticoids, at a mean daily prednisone dose of 20.8 mg (range 10–50). MTX administration began at a dose of 10 mg/week and increased to 20 mg/week. Twelve months after introduction of MTX, six patients were in disease remission and four maintained partial remission on a mean daily prednisone dose of 2 mg. In a second series, three patients were treated by MTX: one maintained remission at 34 months and two relapsed on MTX at 24 months.218,219
Other immunosuppressants. Calcineurin inhibitors (CNI), such as tacrolimus or cyclosporine A, can be used as a steroid-sparing regime in IgG4-related disease patients with contraindications or non-response to other therapies. A target level of 5–7 ng/ml for tacrolimus and one of 80–120 ng/ml for cyclosporine A is documented in the available case reports. There is little evidence for the efficacy of CNI as steroid-sparing agents in IgG4-related disease. One should take into consideration that long-term use of CNI might lead to hypertension or renal insufficiency in older patients.
Cyclophosphamide use has been adapted from the treatment of vasculitis, lupus, and rheumatoid diseases, and administered as an intravenous infusion or tablets 50–100 mg/day, and often for extra-pancreatobiliary disease manifestations. A Chinese controlled trial suggested that cyclophosphamide, in addition to glucocorticoids, lowered the risk of disease relapse (12% at 12 months) compared with glucocorticoids alone (39%), which was, however, associated with increased toxicity. 222
Immunosuppressants display distinctly greater toxicity profiles, accompanied with frequent relapses when used as monotherapy. Too little is known regarding this subject to confidently suggest monotherapy with immunosuppressive agents.
As there has been no randomized controlled study on the treatment of IgG4-related disease, including various drugs, the best evidence-based treatment of this disorder is still unknown. The choice of using certain medication for treatment of IgG4-related disease varies in different countries, among specialties, and different organ involvement. Therefore, multicentre clinical trials with large numbers of patients are needed to define optimal treatment protocols in IgG4-related disease.
Areas of uncertainty and future research
These UEG guidelines set out to provide a rational basis for diagnosing and treating IgG4-related digestive disease. We achieved this goal, however, inherent to a relatively rare disease, there are still several white spots on the map of the digestive tract in conjunction with this enigmatic disease. Consequently, for several areas, evidence is low or possibly non-existent, even if experts agree on a certain practice. Much of this is performed in analogy to other diseases, such as the consideration to treat IRC with ursodeoxycholic acid, as done in other cholangiopathies, such as PBC or PSC. Consequently, as is the case with many other guidelines, areas of future research were identified to provide evidence for future versions. The major topics are the following:
incidence of cancer in patients with IgG4-related digestive disease compared with age-, sex- and risk factor-matched control subjects, and any risk factors for its development should be explored in prospective studies. prospective studies to evaluate the accuracy of imaging modalities (CT, MR, PET-CT) even with the aid of innovative post-processing methods (i.e. Radiomics, Texture Analysis) in the differentiation of focal autoimmune pancreatitis and isolated IRC from pancreatic cancer and cholangiocarcinoma, respectively.
223
as there have been no randomized controlled studies on the treatment of IgG4-related digestive disease, the best evidence-based treatment of this disorder is still unknown. The use of certain medications for treatment of IgG4-related digestive disease varies in different countries, among different specialties, and different organ involvement; therefore, multicentre clinical trials with large numbers of patients are needed to define optimal treatment protocols.
224
IgG4-related digestive disease in children is very rare, and multicentre paediatric studies are necessary for better understanding of the disease course in children, and to define the best treatment choice.
Working party and external expert reviewers
All members of the working party 33 are listed as co-authors. The allocation to the WP is listed in the supplement (Table S2). The following colleagues served as external expert reviewers as part of the working party contributing substantially and are therefore also co-authors of these guidelines in accordance with GUIDE:
Marc G Besselink 1 , Marco J Bruno 2 , László Czakó 3 , Marco del Chiaro 4 , Oleksandra Filippova 5 , Akihisa Fukuda 6 , Sebastien Gaujoux 7 , Phil A Hart 8 , Peter Hegyi 9 , Eduard Jonas 10 , Alisan Kahraman 11 , Alexander Kleger 12 , Olexander Kuryata 5 , Johanna Laukkarinen 13 , Markus M Lerch 14 , Giovanni Marchegiani 15 , Hanns-Ulrich Marschall 16 , Celso Matos 17 , Yair Molad 18 , Dilek Oguz 19 , Aldis Pukitis 20 , Sohei Satoi 21 , John H Stone 22 , Joanne Verheij 23 , Niek de Vries 24
1Department of Surgery, Cancer Center Amsterdam, Amsterdam UMC, University of Amsterdam, Amsterdam, the Netherlands
2Department of Gastroenterology and Hepatology; Erasmus Medical Center, University Medical Center, Rotterdam, the Netherlands
3First Department of Medicine, University of Szeged, Szeged, Hungary
4Division of Surgical Oncology, Department of Surgery, University of Colorado Anschutz Medical Campus, Denver, USA, CO Division of Surgery, CLINTEC-Karolinska Institutet, Stockholm, Sweden, Division of Surgical Oncology, Department of Surgery, Johns Hopkins University, Baltimore, USA
5Internal Medicine 2 and Physiology, Dnipropetrovsk State Medical Academy, Dnipro, Ukraine
6Department of Gastroenterology & Hepatology; Kyoto University Graduate School of Medicine, Japan
7Department of Pancreatic and Endocrine Surgery, Cochin Hospital, Paris, APHP, France
Université de Paris, France
8Division of Gastroenterology, Hepatology, and Nutrition, The Ohio State University Wexner Medical Center, Columbus, Ohio, USA
9Institute for Translational Medicine, Szentágothai Research Centre, Medical School, University of Pécs, Pécs, Hungary
10Surgical Gastroenterology Unit, Department of Surgery, University of Cape Town and Groote Schuur Hospital, Cape Town, South Africa
11Department of Gastroenterology and Hepatology, University Clinic of Essen, Germany
12University Medical Center Ulm, Center for Internal Medicine, Department of Internal Medicine I, University of Ulm, Ulm, Germany
13Department of Gastroenterology and Alimentary Tract Surgery, Tampere University Hospital, and Faculty of Medicine and Health Technology, Tampere University, Finland
14Department of Medicine A, University Medicine Greifswald, Greifswald, Germany
15Department of Surgery, Pancreas Institute, University and Hospital Trust of Verona, Verona, Italy
16Department of Molecular and Clinical Medicine/Wallenberg Laboratory, University of Gothenburg, Gothenburg, Sweden
17Radiology Department, Champalimaud Centre for the Unknown, Lisbon, Portugal
18Institute of Rheumatology, Rabin Medical Center, Beilinson Hospital, and The Laboratory of Inflammation Research, Felsenstein Medical Research Center, Sackler Faculty of Medicine, Tel Aviv University, Petach Tikva, Israel
19Department of Gastroenterology, Kırıkkale University School of Medicine, Kırıkkale, Turkey
20Center of Gastroenterology, Hepatology and Nutrition, Pauls Stradins
Clinical University Hospital; University of Latvia, Riga, Latvia
21Division of Pancreatobiliary Surgery, Department of Surgery, Kansai Medical University, Japan
22Harvard Medical School The Edward A. Fox Chair in Medicine Massachusetts General Hospital, Boston, MA, USA
23Department of Pathology, Cancer Center Amsterdam, Amsterdam UMC, University of Amsterdam, Amsterdam, the Netherlands
24Department of Clinical Immunology & Rheumatology, Academic Medical Center, University of Amsterdam, Amsterdam, the Netherlands
Online
Parts of these guidelines, are available online (
Disclaimer
These clinical practice guidelines (CPGs) are developed to assist clinicians with decisions about appropriate health care in patients with IgG4-related disease. They detail the assessment and management of many common (and some rare but important) conditions and have been developed for use in healthcare settings. As such, these CPGs are targeted at clinicians only. Patients or other community members using these CPGs should do so in conjunction with a health professional and should not rely on the information in these guidelines as professional medical advice.
These CPGs do not constitute a textbook and therefore deliberately provide little, if any, explanation or background to the conditions and treatment outlined. They are however designed to rapidly acquaint the reader with the clinical problem and provide practical advice regarding assessment and management.
These CPGs are developed by a multidisciplinary team of practising clinicians by consensus and based on the evidence available.
The recommendations contained in these CPGs do not indicate an exclusive course of action or standard of care. They do not replace the need for application of clinical judgment to each individual presentation, nor variations based on locality and facility type.
The inclusion of links to external websites does not constitute an endorsement of those websites nor the information or services offered.
The authors of these CPGs have made considerable efforts to ensure the information upon which they are based is accurate and up to date. Users of these CPGs are strongly recommended to confirm that the information contained within them, especially drug doses, is correct by way of independent sources. The authors accept no responsibility for any inaccuracies, information perceived as misleading, or the success or failure of any treatment regimen detailed in the CPGs.
Supplemental Material
sj-pdf-1-ueg-10.1177_2050640620934911 - Supplemental material for European Guideline on IgG4-related digestive disease – UEG and SGF evidence-based recommendations
Supplemental material, sj-pdf-1-ueg-10.1177_2050640620934911 for European Guideline on IgG4-related digestive disease – UEG and SGF evidence-based recommendations by J-Matthias Löhr, Ulrich Beuers, Miroslav Vujasinovic, Domenico Alvaro, Jens Brøndum Frøkjær, Frank Buttgereit, Gabriele Capurso, Emma L Culver, Enrique de-Madaria, Emanuel Della-Torre, Sönke Detlefsen, Enrique Dominguez-Muñoz, Piotr Czubkowski, Nils Ewald, Luca Frulloni, Natalya Gubergrits, Deniz Guney Duman, Thilo Hackert, Julio Iglesias-Garcia, Nikolaos Kartalis, Andrea Laghi, Frank Lammert, Fredrik Lindgren, Alexey Okhlobystin, Grzegorz Oracz, Andrea Parniczky, Raffaella Maria Pozzi Mucelli, Vinciane Rebours, Jonas Rosendahl, Nicolas Schleinitz, Alexander Schneider, Eric FH van Bommel, Caroline Sophie Verbeke, Marie Pierre Vullierme, Heiko Witt and the UEG guideline working group in United European Gastroenterology Journal
Footnotes
Acknowledgements
We thank both the Swedish Society for Gastroenterology (SGF) and the Dutch Society for Gastroenterology for their readiness to serve as national societies of UEG supporting these guidelines. Furthermore, the support of the European Pancreatic Club and the United European Gastroenterology is acknowledged for providing space and time for the face-to-face meetings in the development of these guidelines.
Conflict of interest
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: AL reports speaker fees from Bracco, GE Healthcare, Merck, Bayer and Bristol-Myers-Squibb; DGD received fellowship grant from Gilead Sciences; DA received research support from Intercept Pharma and Vesta and speaker fees from Intercept Pharma and Aboca; ELC reports consultation fees from Xencor pharmaceuticals; MPV reports honoraria from Guerbet; JML reports research support from Mylan and lecture fees from Abbott and Mylan; MV reports research support from Mylan and lecture fees from Abbott and Mylan, NS reports honoraria and consultation fees from Baxalta, Shire, LFB, CSL Behring and Novartis; UB reports research support from Falk and Intercept, honoraria and consultation fees from Intercept and lecture fees from Abbvie, Falk Foundation and Intercept. The other authors have nothing to disclose regarding the work under consideration for publication.
Funding
We gratefully acknowledge the support from the National Societies Committee of the United European Gastroenterology (UEG) for the conduct of these guidelines independent from other sources. No other funding was received.
ORCID iDs
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.