
Abstract
Background
The relationship between vedolizumab trough levels and combined endoscopic and clinical remission is unknown.
Objective
To compare vedolizumab trough levels in patients with and without combined remission within the first year of treatment.
Methods
We prospectively collected vedolizumab trough levels in 51 consecutive patients (28 Crohn's disease (CD) and 23 ulcerative colitis (UC)) before all infusions up to week 22, and at weeks 38 and 54, with concentrations measured after study completion. Centrally read endoscopy was performed at a median of 46 weeks. The primary outcome was combined endoscopic (CD: Simple endoscopic score for CD (SES-CD) < 4 without ulceration; UC: Mayo endoscopic subscore ≤ 1) and clinical remission (CD: resolution of abdominal pain; UC: resolution of rectal bleeding; both: resolution of altered bowel habit).
Results
Median vedolizumab trough levels at weeks 6 (25.7 vs 15.6 µg/mL; P = 0.015) and 22 (15.1 vs 4.9 µg/mL; P = 0.001) were higher in patients with combined remission. A threshold of 22 µg/mL at week 6 (area under the curve (AUC) 0.733; 95% confidence interval 0.567–0.899) and 8 µg/mL at week 22 (AUC 0.819; 95% confidence interval 0.692–0.946) predicted combined remission.
Conclusion
Early vedolizumab trough levels predicted combined endoscopic and clinical remission highlighting their possible use in clinical practice.
Key summary
Summarize the established knowledge on this subject
An association between vedolizumab trough levels and clinical or endoscopic outcomes in isolation has been shown. The relationship between vedolizumab trough levels and combined, endoscopic and clinical remission, as mandated by the Selecting Therapeutic Targets in Inflammatory Bowel Disease programme, is unknown. Induction and early maintenance vedolizumab trough levels are associated with combined, endoscopic and clinical remission. Only 4% of patients with week 6 vedolizumab trough levels below 17.0 µg/mL achieved combined remission by week 54. Early therapeutic drug monitoring could help identify patients at risk of unfavourable outcomes and provide an early opportunity for therapeutic intervention (e.g. dosing-interval shortening)
What are the significant or new findings of this study?
Introduction
Vedolizumab is a humanized monoclonal antibody against α4β7 integrin, which prevents mucosal addressin cell adhesion molecule-1-mediated lymphocyte homing to the gut. 1 It is effective as induction and maintenance therapy for patients with moderate to severe Crohn's disease (CD) and ulcerative colitis (UC).2–4
Recently, a combination of patient-reported outcomes (PROs) and objective measures of disease has been proposed as the preferred endpoint for clinical trials.5,6 Identifying early predictors of combined endoscopic and PRO remission in a drug with a delayed onset of maximal effect, such as vedolizumab, 7 would be valuable to avoid futile costly treatment and to seize early opportunities to change the therapeutic strategy. Vedolizumab trough levels during induction and maintenance have been shown to be associated with endoscopic8,9 and clinical9–11 outcomes, respectively. Nevertheless, published real-life studies have not yet focused on the relationship between vedolizumab trough levels and combined endoscopic and clinical remission, as defined by the Selecting Therapeutic Targets in Inflammatory Bowel Disease (STRIDE) programme.
The principal aim of our study was to explore the predictive value of early vedolizumab trough levels for endoscopic and clinical remission by the end of the first year of treatment.
Methods
Patient population, study design and outcomes
This was a prospective observational study of consecutive inflammatory bowel disease (IBD) patients who started vedolizumab treatment at a single tertiary referral centre between May 2016 and June 2017. The study design conforms with the 1975 Declaration of Helsinki, was approved by the National Committee of Medical Ethics (0120-013/2016-2; KME 18 January 2016) and all patients provided written informed consent.
All consecutive patients with IBD treated with vedolizumab at our centre were included in this study. The decision to commence vedolizumab was taken by a multidisciplinary team, based on clinical, biochemical and endoscopic evidence of disease activity. All included patients underwent endoscopy within 3 months prior to starting vedolizumab and were proven to have active disease. Patients who underwent endoscopy later than week 54 after starting vedolizumab were excluded from the study.
Vedolizumab levels were measured at trough (i.e. just before the infusion) at weeks 2, 6, 10, 14, 22, 38 and 54 after starting treatment. For patients who discontinued treatment during the study due to drug failure, trough levels were analysed until their last infusion.
Mucosal healing was assessed by a complete colonoscopy between weeks 20 and week 54. In patients with dosing-interval shortening, colonoscopy was performed after at least two infusions at a shortened interval. Patients who underwent surgery had unequivocally active disease based on clinical and biochemical data, and cross-sectional imaging (in CD) or flexible sigmoidoscopy (in UC), and thus did not undergo a complete colonoscopy; they were considered to have no mucosal healing. Colonoscopies were recorded, the recordings anonymized and then assessed centrally by an expert endoscopist (NSe) who was blinded to the patients' condition. Endoscopic remission was defined as a Simple endoscopic score for CD (SES-CD) of < 4, 12 and the absence of any mucosal ulceration (in CD) or a Mayo endoscopic subscore of ≤ 1 (in UC). 13
Clinical remission was defined as resolution of abdominal pain and altered bowel habit for CD, and resolution of rectal bleeding and altered bowel habit for UC. 6 We used two-item PRO scores with remission defined as mean daily stool frequency of ≤ 1.5 and abdominal pain ≤ 1 for CD, 14 and a rectal bleeding score of 0 and a stool frequency score of ≤ 1 for UC, 15 both without concomitant steroids. Combined remission was defined as endoscopic remission and concomitant clinical remission at the end of follow-up.
Clinical management of patients
Intravenous infusions of vedolizumab (300 mg) were administered at 0, 2, 6 and 10 weeks (induction), and then from week 14 onwards once every 4 or 8 weeks (maintenance). Thiopurines were discontinued during induction and by week 10 at the latest. Both patients with CD and UC (off-label) were eligible to receive an additional infusion at week 10. The decision for an additional infusion at week 10 and/or for dosing-interval shortening to every 4 weeks after week 14 was made by the treating physician in case of inadequate response judged clinically (liquid stools and/or blood in stools) and by biomarkers (elevated C-reactive protein and/or elevated faecal calprotectin). The time to dosing-interval shortening was defined as the week when the patient first received the infusion at the shorter interval. All treating physicians were blinded to vedolizumab trough level measurements, which thus did not influence management.
Vedolizumab trough levels and antibodies to vedolizumab
Vedolizumab concentrations were measured using the Conformité Européenne-marked apDia vedolizumab enzyme-linked immunosorbent assay (Turnhout, Belgium) with a measurement range between 1–50 µg/mL. Antibodies against vedolizumab were measured with a drug-sensitive bridging assay. 16
Statistical analysis
All statistical analyses were performed using SPSS, version 22 (IBM, Chicago, USA). Descriptive statistics were given as percentages for nominal variables, and as median and interquartile ranges (IQRs) for continuous variables. The χ 2 -test was used to analyse nominal variables and the Mann–Whitney U-test for continuous unpaired variables. Cut-off vedolizumab trough levels to predict combined remission were identified using receiver operating curve analysis with the calculation of Youden's J statistic. 17 Stepwise binary logistic regression was performed to identify independent variables associated with combined remission. To avoid introducing bias by excluding patients who did not undergo endoscopy within 54 weeks, an intention-to-treat analysis was performed by including their outcome data based on endoscopy performed after week 54. P < 0.05 was considered statistically significant.
Results
Patient characteristics
Patient demographic and clinical characteristics.
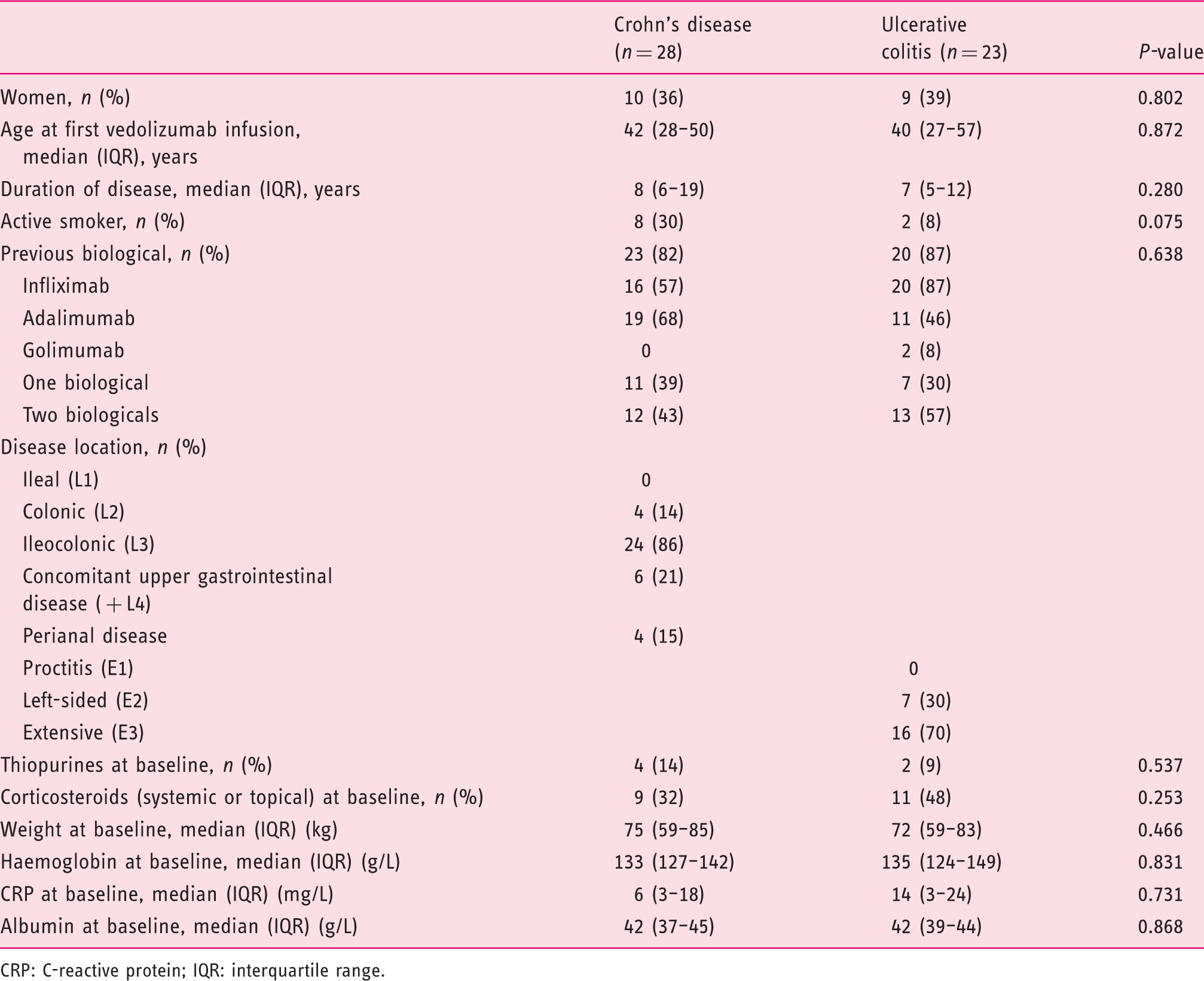
CRP: C-reactive protein; IQR: interquartile range.
Treatment course
In the overall study population, 13 patients (10 CD and 3 UC; 25%) received an additional infusion of vedolizumab at week 10. Dosing-interval shortening was undertaken in 23 patients (15 CD and 8 UC; 45%). The median time to dose optimization was 25 weeks (IQR 18–34). Of 51 patients, 36 (71%) remained on treatment at week 54. The discontinuation rate was similar for CD (8/28; 29%) and UC (7/23; 30%) (P = 0.884). The median time to discontinuation was 34 weeks (IQR 20–54).
Endoscopic, clinical and combined remission
A total of 45 patients (26 CD and 19 UC) were assessed with colonoscopy, 6 (2 CD and 4 UC) underwent surgery and were considered to have no mucosal healing. The median time to endoscopy was 46 weeks (IQR 31–54). By week 54, a total of 19 patients (37%) achieved mucosal healing. The rate of mucosal healing was numerically higher in UC (11/23; 48%) than in CD (8/28; 29%) (P = 0.157) patients. In CD patients, the median SES-CD for patients with mucosal healing was 1 (IQR 0–3); in the 11 UC patients with mucosal healing, 9 had an endoscopic Mayo score of 0 and 2 of 1.
By the end of follow-up, 26 patients (51%) achieved clinical remission with no difference between the two diseases (CD 16/28; 57% vs UC 10/23; 43%; P = 0.331).
Combined endoscopic and clinical remission was achieved in 16 patients (31%) with no difference between the two diseases (CD 7/28; 25% vs UC 9/23; 39%; P = 0.279). Combined remission was achieved in 3/23 patients (13%) who underwent dosing-interval shortening.
Vedolizumab trough levels and anti-vedolizumab antibodies
Median vedolizumab trough levels in the overall population during induction were 21.5 µg/mL (IQR 16.2–29.7) at week 2, 17.3 µg/mL (IQR 10.0–26.7) at week 6 and 17.0 µg/mL (IQR 8.5–22.3) at week 10. During maintenance, median trough levels decreased to 9.3 µg/mL (IQR 3.9–15.9) at week 14, 7.6 µg/mL (IQR 3.4–15.1) at week 22, 9.4 µg/mL (IQR 5.0–14.1) at week 38 and 10.2 µg/mL (IQR 4.6–16.1) at week 54. The median trough levels at weeks 14 (13.1 vs 8.8 µg/mL; P = 0.612) and 22 (8.3 vs 7.6 µg/mL; P = 0.993) in patients who received an additional infusion at week 10 did not differ from those who did not receive it. Trough levels in patients who were treated with thiopurines at inclusion did not differ from those of the rest of the cohort.
Median vedolizumab trough levels and combined endoscopic and clinical remission by week 54 of vedolizumab treatment.
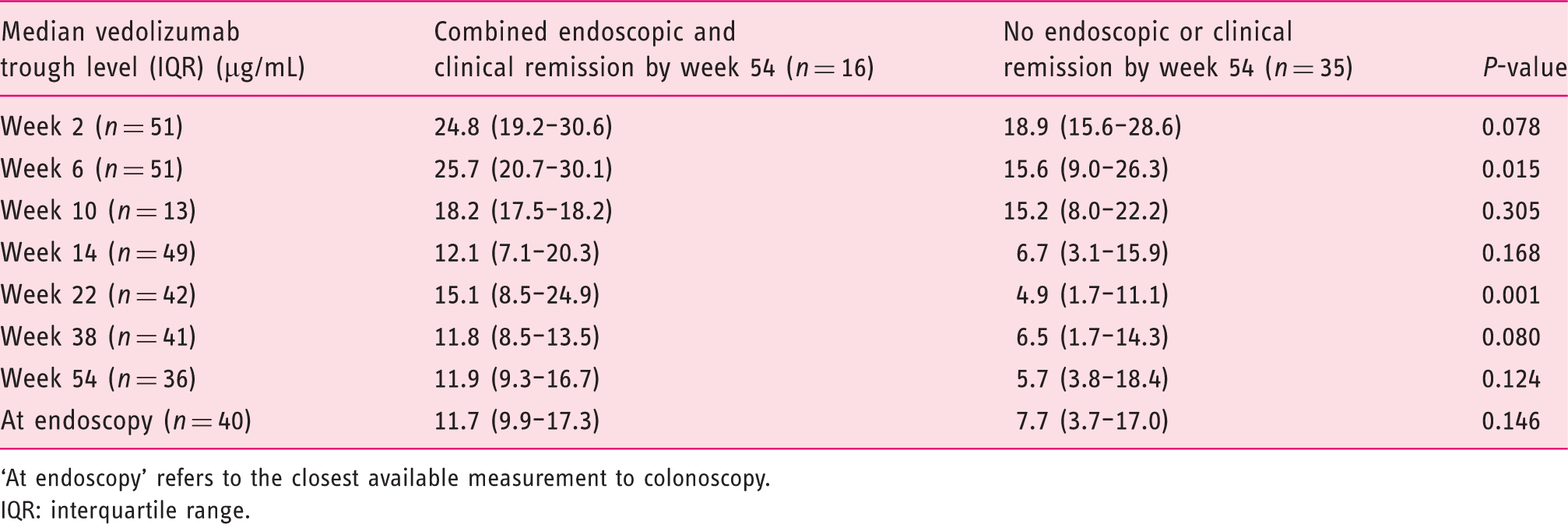
‘At endoscopy’ refers to the closest available measurement to colonoscopy.
IQR: interquartile range.
Variables associated with combined endoscopic and clinical remission by week 54 of vedolizumab treatment.
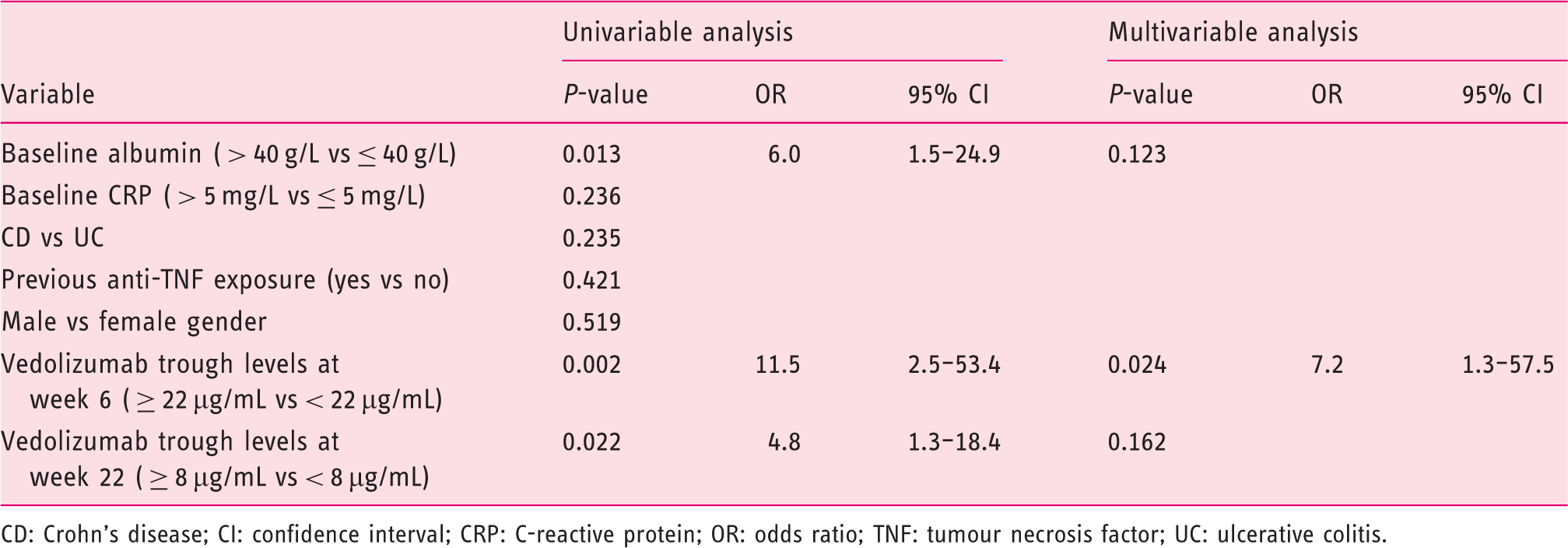
CD: Crohn's disease; CI: confidence interval; CRP: C-reactive protein; OR: odds ratio; TNF: tumour necrosis factor; UC: ulcerative colitis.
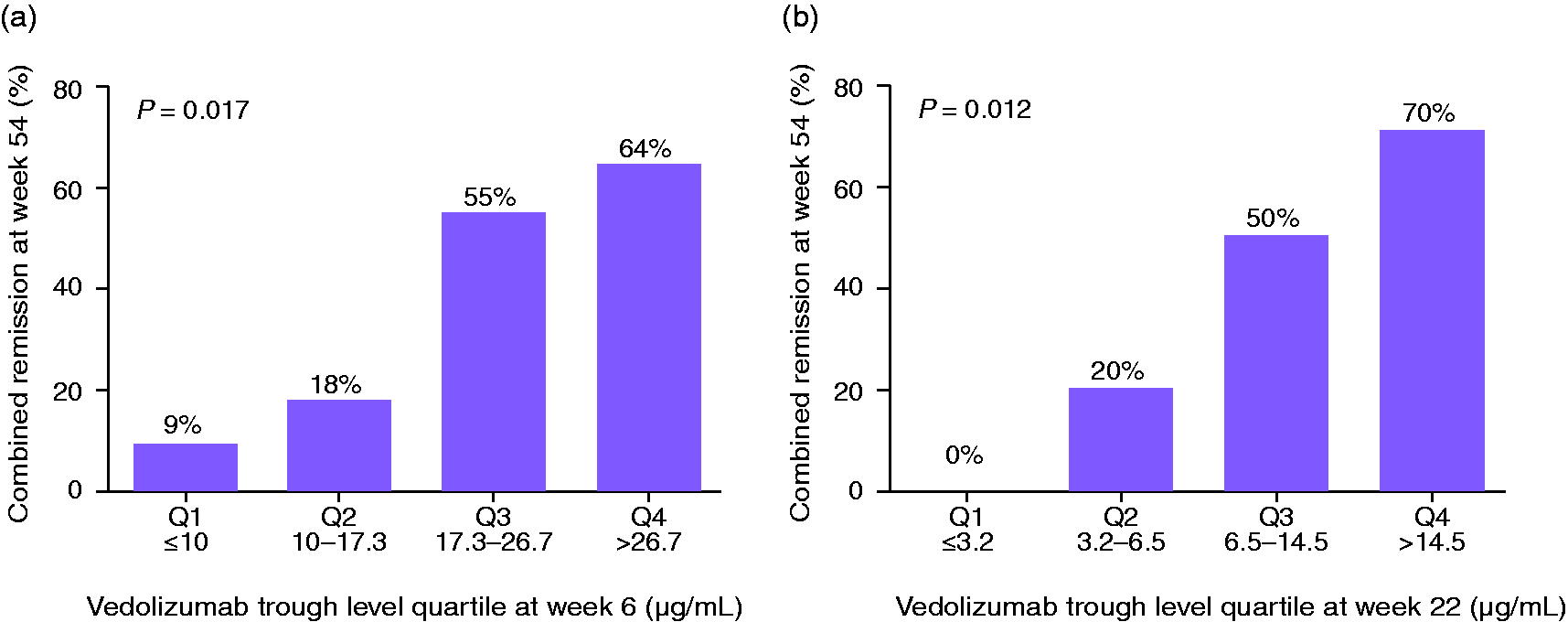
In the overall population, a week 6 trough level of 22.0 µg/mL predicted combined remission with a sensitivity, specificity, positive predictive value and negative predictive value of 77, 73, 72 and 88%, respectively (area under the curve (AUC) 0.733; 95% confidence interval (CI) 0.567–0.899) (Figure 1(a)). Among patients with week 6 trough levels below 17.0 µg/mL, 1/24 (4%) achieved combined remission. At week 22, a trough level of 8.0 µg/mL predicted combined remission with a sensitivity, specificity, positive predictive value and negative predictive value of 79, 75, 65 and 86%, respectively (AUC 0.819; 95% CI 0.692–0.946) (Figure 1(b)). Among patients with week 22 trough levels < 4 µg/mL, 1/12 (8%) achieved combined remission (Figure 2). There was no difference between the per-protocol and intention-to-treat analysis (using outcomes for the four patients who underwent endoscopy after week 54–weeks 66–72).
Receiver operating characteristic curve for vedolizumab trough levels at week 6 (a) and week 22 (b) to predict combined endoscopic and clinical remission by week 54 of vedolizumab treatment. Percentage of patients with combined, endoscopic and clinical remission by vedolizumab trough level at weeks 6 (a) and 22 (b).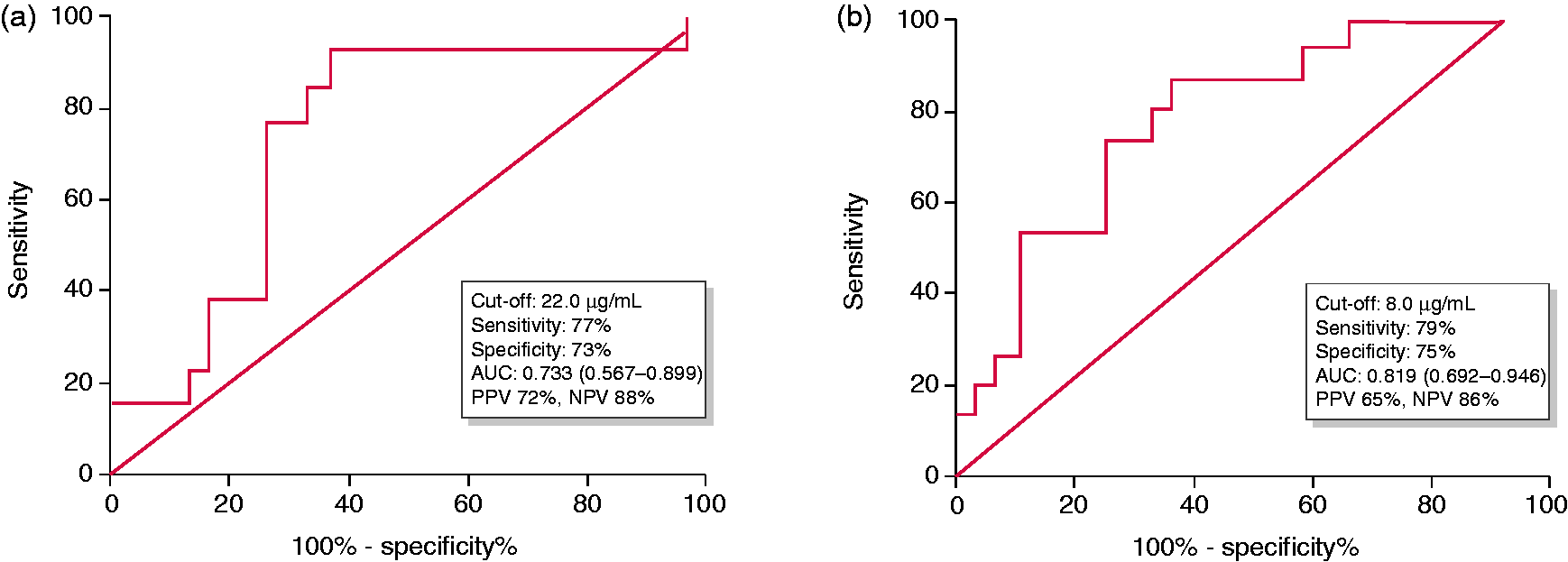
Vedolizumab was undetectable in 11 samples from seven patients (four CD and three UC). None of them achieved combined remission despite dosing-interval shortening; undetectable levels were measured at a median of week 30 (IQR 22–38). Median week 6 trough levels in this subgroup were lower compared to patients with detectable trough levels (7.4 vs. 21.7 µg/mL; P = 0.005). No antibodies to vedolizumab were detected using a drug-sensitive assay.
Factors associated with combined remission
Univariable analysis identified baseline albumin > 40 g/L and vedolizumab trough levels ≥ 22 µg/mL at week 6, and ≥ 8 µg/mL at week 22, as variables associated with combined remission (Table 3). Only vedolizumab trough levels ≥ 22 µg/mL at week 6 remained statistically significant by multivariable analysis (odds ratio 7.2; 95% CI 1.3–57.5; P = 0.024).
Discussion
The main new finding of our study is the predictive value of induction and early maintenance vedolizumab trough levels for robust combined endoscopic and clinical remission by the end of the first year of vedolizumab treatment. Apart from confirming an exposure–response relationship for a stringent endpoint, we were able to identify patients during induction who ultimately achieved favourable outcomes and those who have a low probability of combined remission using early vedolizumab trough levels. This offers an opportunity for intervention (dosing-interval shortening or treatment change) in patients with a low likelihood of success with standard dosing.
An important finding of our study was the 96% negative predictive value of the vedolizumab trough level threshold of 17 µg/mL at week 6 for combined endoscopic and clinical remission. Trough levels below this threshold were observed in a high proportion of our patients. Identifying these thresholds is crucial to be able to select patients who could benefit from dosing-interval shortening or possible alternative strategies to optimize treatment. 18 Similar thresholds for unfavourable outcomes have been recognized by others. In patients with UC with week 6 trough levels < 17.1 µg/mL, the rate of endoscopic remission was 20.1%. 10 In a mixed population, trough levels < 18.5 µg/mL at week 6 predicted the need for dosing-interval shortening, but only clinical outcomes were studied. 19
Week 6 vedolizumab trough levels have already been identified as being predictive of mucosal healing with a threshold of 18 µg/mL. 8 A subsequent study identified a week 6 threshold of > 24 µg/mL as being associated with attaining clinical, biochemical and endoscopic therapeutic targets. 9 These thresholds are broadly in line with our threshold of 22 µg/mL, which also adequately predicted combined remission. The observed differences in thresholds may reflect minor differences in outcome definitions, the timing of endoscopic assessment and the assay used. All studied cohorts had a high proportion of patients who previously failed anti-TNF agents.
Despite a persistent numerical trend for maintenance time points beyond week 22, differences in our study were not statistically significant, possibly owing to decreasing statistical power as patients discontinued treatment. In registration trials, no convincing relationship was demonstrated between week 46 trough levels and clinical remission at week 52. 2 In contrast to the positive relationship between maintenance trough levels and endoscopic remission that has been shown for infliximab,20,21 vedolizumab trough levels measured closest to endoscopy in our study were only marginally higher in patients with endoscopic remission. Taken together with currently available studies, 22 these data indicate that trough level measurements during maintenance currently do not appear to be strongly associated with outcomes.
In our study, a high percentage of patients required dosing-interval shortening due to inadequate response. The efficacy of this approach was limited in achieving combined remission. In a recent meta-analysis, the pooled efficacy rate of dosing-interval shortening was 53.8%; however, response was not evaluated endoscopically. 23 In two studies where endoscopic outcomes of patients undergoing dosing-interval shortening were presented separately, 438 and 31% 9 achieved mucosal healing. A substantially higher percentage, 56%, achieved clinical remission in the latter study where both outcomes were assessed.
Patients receiving vedolizumab infusions every 4 weeks had remarkably low trough levels before dosing-interval shortening compared to data from registrational and subsequent studies, 24 possibly reflecting different patient characteristics. Interestingly, these patients in our study failed to achieve combined remission after dosing-interval shortening, despite reaching trough levels associated with combined remission in patients who reached this endpoint without interval shortening to every 4 weeks at matched time points. In the only available report on the relationship between changes in trough levels after dosing-interval shortening and clinicobiological outcomes, responders and non-responders had similar baseline trough levels, but the former exhibited a numerically larger increase in trough levels. 25
The biological plausibility of measuring vedolizumab levels has been questioned because full saturation of the α4β7 integrin has been consistently achieved at drug concentrations of 1–3 µg/mL. 11 Additional mechanisms involving innate immunity without significant changes in intestinal T cell abundance or T cell receptor profile have recently been outlined. 26 Furthermore, Boden et al. have highlighted differences between responders and non-responders: the former maintained full saturation at lower concentrations than the latter. 27
The strengths of our study are the prospective design in a real-life clinical setting with central reading of endoscopies and the use of the strict primary outcome of combined remission. The main limitations of our study are the single-centre design and the small sample size of our cohort. The study was not adequately powered for subgroup analysis (e.g. CD vs. UC; dosing every 4 weeks vs. every 8 weeks). The majority of patients had failed previous anti-TNF treatment, which prevents the extrapolation of our findings to biologically naïve patients. Owing to its excellent safety profile, vedolizumab is likely to become the biological agent of choice in elderly patients, who were underrepresented in our population. Distinction between primary non-response and secondary loss of response was not done in our study, as we decided to conduct only one endoscopy. To unequivocally distinguish primary from secondary non-response, we would need to conduct an additional endoscopy after induction. The low rate of combined remission with dosing-interval shortening precluded further pharmacokinetic analysis in this subgroup.
In conclusion, our prospective real-life observational study with centrally evaluated endoscopy identified vedolizumab trough levels at week 6 to be a predictor of combined endoscopic and clinical remission, as defined by the STRIDE consensus. Taken together with the findings of others, trough levels at week 6 appear to be a promising tool to assist clinical decisions. Our hypothesis-generating study warrants further research to explore whether patients with a low likelihood of combined remission (i.e. those with week 6 trough levels < 17 µg/mL) could benefit from early dosing-interval shortening, possibly including other relevant patient-reported outcomes.
Supplemental Material
Supplemental material for Early vedolizumab trough levels predict combined endoscopic and clinical remission in inflammatory bowel disease
Supplemental Material for Early vedolizumab trough levels predict combined endoscopic and clinical remission in inflammatory bowel disease by Jurij Hanžel, Nejc Sever, Ivan Ferkolj, Borut Štabuc, Nataša Smrekar, Tina Kurent, Matic Koželj, Gregor Novak, Griet Compernolle, Sophie Tops, Ann Gils and David Drobne: on behalf of Observatoire Francophone de la Sclérose en Plaques (OFSEP), Société Francophone de la Sclérose en Plaques (SFSEP), the Radiologically Isolated Syndrome Consortium (RISC) and the Pediatric Radiologically Isolated Syndrome Consortium (PARIS) in United European Gastroenterology Journal
Footnotes
Acknowledgements
We would like to thank Carmen Bobnar Sekulić and Tadeja Polanc for their help collecting samples, and their administrative support. Part of this work was presented at the European Crohn’s and Colitis Organisation Congress, Vienna 2018; Digestive Disease Week, Washington DC 2018; and the United European Gastroenterology Week, Vienna 2018.
JH, DD and AG designed the study. DD supervised the study. JH and NSe analysed the data, JH and DD interpreted the data and prepared the manuscript. IF, BS, NSm, TK, MK, GN, GC and ST acquired the data. All authors critically reviewed the manuscript and approved the final submitted draft.
Declaration of conflicting interests
JH reports lecture fees from Biogen and Takeda outside the submitted work. IF reports lecture and consultant fees from MSD, Abbvie, Takeda and Krka outside the submitted work. BŠ reports lecture and consultant fees from Krka, Bayer and Takeda, and grants from Krka outside the submitted work. NSm reports lecture fees from Takeda outside the submitted work. GN reports lecture and consultant fees from Takeda, MSD and Abbvie. MK reports lecture fees from Takeda outside the submitted work. AG reports lecture fees from MSD, Janssen Biologicals, Pfizer, Takeda, Novartis and Abbvie, consultant fees from Takeda and research grant support from Pfizer, MSD and Takeda, all outside the submitted work. KU Leuven holds a license agreement with R-biopharm, apDia and Merck. DD reports speaker and consultant fees from MSD, Abbvie, Takeda, Pfizer, Janssen, Krka, Dr. Falk Pharma and Ferring, all outside the submitted work. All other authors have nothing to disclose.
Ethics approval
The study design conforms to the 1975 Declaration of Helsinki, was approved by the National Committee of Medical Ethics (0120-013/2016-2; KME 18 January 2016).
Funding
There were no external funding sources or sponsoring societies for this study.
Informed consent
All patients provided written informed consent for inclusion in the study.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.