
Abstract
Chronic pain affects a large part of the global population, leading to an increase of opioid use. Opioid-induced constipation (OIC), a highly prevalent adverse effect of opioid use, has a major impact on patients’ quality of life. Thanks to the introduction of new drugs for chronic constipation, which can also be used in OIC, and the development of peripherally acting mu-opioid receptor blockers, specifically for use in OIC, therapeutic options have seen major development. This review summarises current and emerging treatment options for OIC based on an extensive bibliographical search. Efficacy data for laxatives, lubiprostone, prucalopride, linaclotide, oxycodone/naloxone combinations, methylnaltrexone, alvimopan, naloxegol, naldemedine, axelopran, and bevenopran in OIC are summarised.
Keywords
Introduction
Chronic pain, which affects 20% of the European population, is prevalent in chronic non-malignant conditions such as low back pain, migraine, fibromyalgia and many other disorders.1,2 The financial burden of chronic pain to society is considerable and was estimated at €200 billion in Europe and $150 billion in the United States (US) yearly. 3 Successful management of chronic pain is of great importance to reduce life-limiting effects on work, physical and emotional well-being, and many other aspects concerning quality of life (QoL). Despite advances in chronic pain management, up to 90% of patients with chronic pain are taking opioids. 4 These opioid agonists have a well-established role in acute pain management, but their role in chronic pain management is controversial. 4
Definition of opioid-induced constipation according to the Rome IV criteria.

In line with an increasing unmet need of OIC treatments, recently several new therapeutic approaches were evaluated. In this manuscript, we review the current state of knowledge and recent advances in the management of OIC in adults.
Pathophysiology
The pathophysiology underlying OIC is complex and mainly reflects actions of opioids on opioid receptors in the GI system. Three major types of opioid receptors are present in the human enteric nervous system: mu-, kappa- and delta-opioid receptors, all G-protein–coupled receptors.8,9 Opioid receptor activation hyperpolarises enteric neurons through inhibition of calcium channels and activation of potassium channels, thus generating reduced action potential firing and decreased neurotransmitter release within the enteric nervous system.
10
Mu-opioid receptors, expressed mostly on myenteric and submucosal neurons throughout the GI tract, are predominantly responsible for the inhibition of propulsive motility by opioids and other adverse effects.
11
This propulsive motility is controlled by the myenteric plexus causing contractions and relaxations of longitudinal and circular muscle layers. Actions of interneurons coordinate this activity, determining muscle response timing. Triggers to induce contractile patterns, such as peristalsis, are driven through activation of intrinsic sensory neurons, of which cell bodies can be located in the myenteric or submucosal ganglia.
12
Figure 1 summarises receptors and modes of action of agents used to treat OIC.
Summary figure of receptors and modes of action of agents used to treat opioid-induced constipation. (1): Osmotic laxatives. (1) Stimulant laxatives. (3) Lubiprostone. (4). Linaclotide. (5). Prucalopride. (6). Naloxone and peripherally acting mu-opioid receptor antagonists. 5-HT4: 5-hydroxytryptamine receptor 4; Ach: acetylcholine; ATP: adenosine triphosphate; cGMP: cyclic guanosine monophosphate; GTP: guanosine triphosphate; NDP: nucleoside diphosphate kinase; Sub P: substance P; VIP: vasoactive intestinal polypeptide.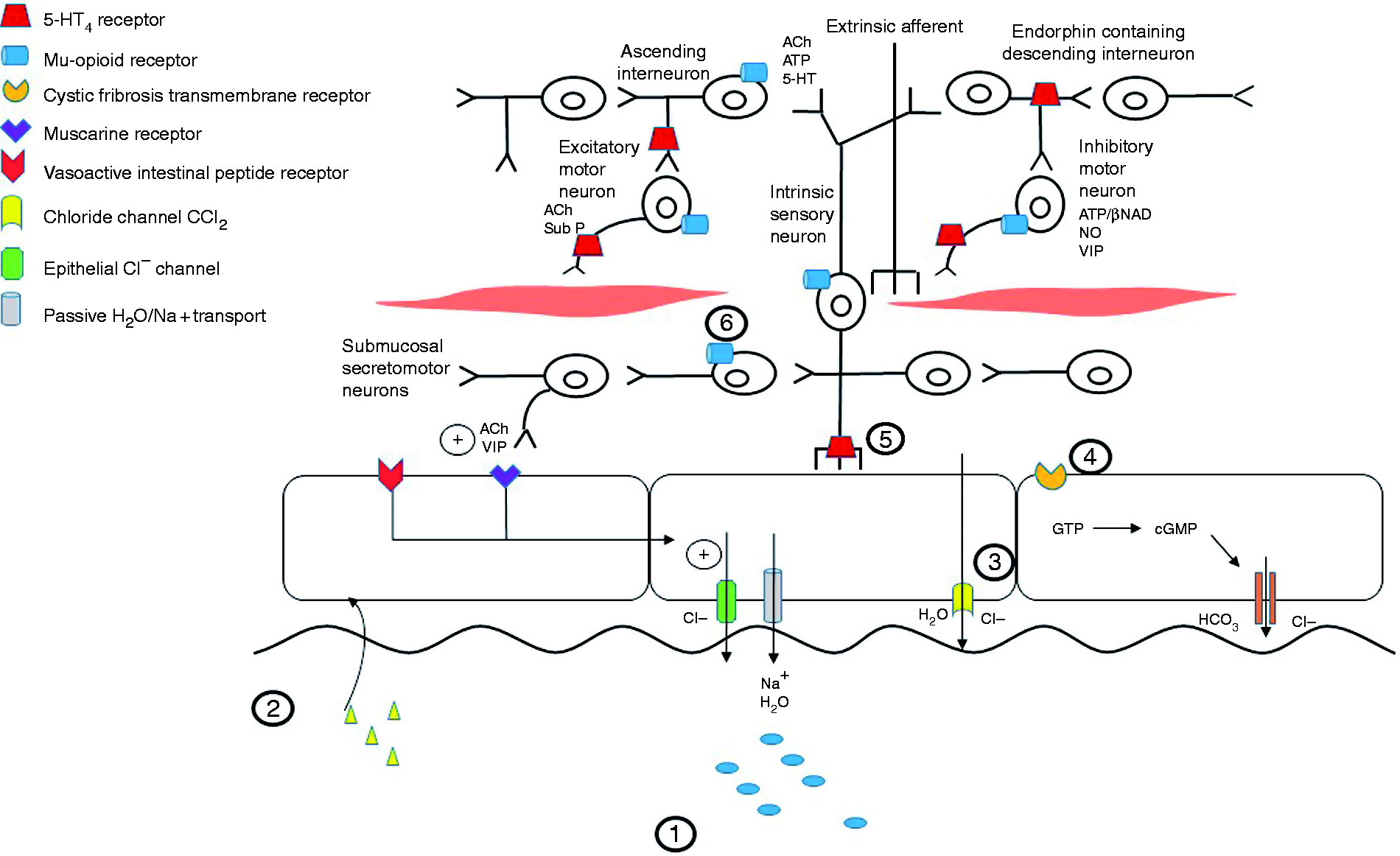
Beside inhibition of GI peristalsis, opioids increase resting tone of sphincters, non-propulsive activity and spasms. 11 These effects are all caused by neuro-neuronal and neuromuscular transmission inhibition through binding of opioid agonists on mu- and delta-opioid receptors, inhibiting acetylcholine release and purine/nitric oxide release from enteric interneurons and inhibitory motor neurons, respectively. 10 Finally, opioid agonists binding on mu and delta receptors on submucosal secretomotor neurons inhibits chloride secretion and osmotic luminal water movement, resulting in reduced colonic water content and dry, hard stools (Figure 1). 10
Current treatment
Laxatives
Laxatives can improve constipation through a number of actions: They can increase colonic residue to stimulate peristalsis (bulk laxatives), attract water by an osmotic gradient (osmotic laxatives), soften stool consistency through direct effects on faecal mass (stool softeners), and stimulate intestinal motility by acting on enteric nerves and increasing secretions by decreasing colonic absorption (stimulant laxatives).
9
Figure 1 summarises the mode of action of treatments used for OIC. Figure 2 gives an overview of the results of the randomised, controlled trials performed in OIC patients.
Overview of randomised, controlled trials for opioid-induced constipation in non-cancer patients, with corresponding responder definitions. *p < 0.05. PAC-SYM: patient assessment of constipation symptoms; QD: once daily; QOD: once every other day; RFBM: rescue-free bowel movement; SBM: spontaneous bowel movement.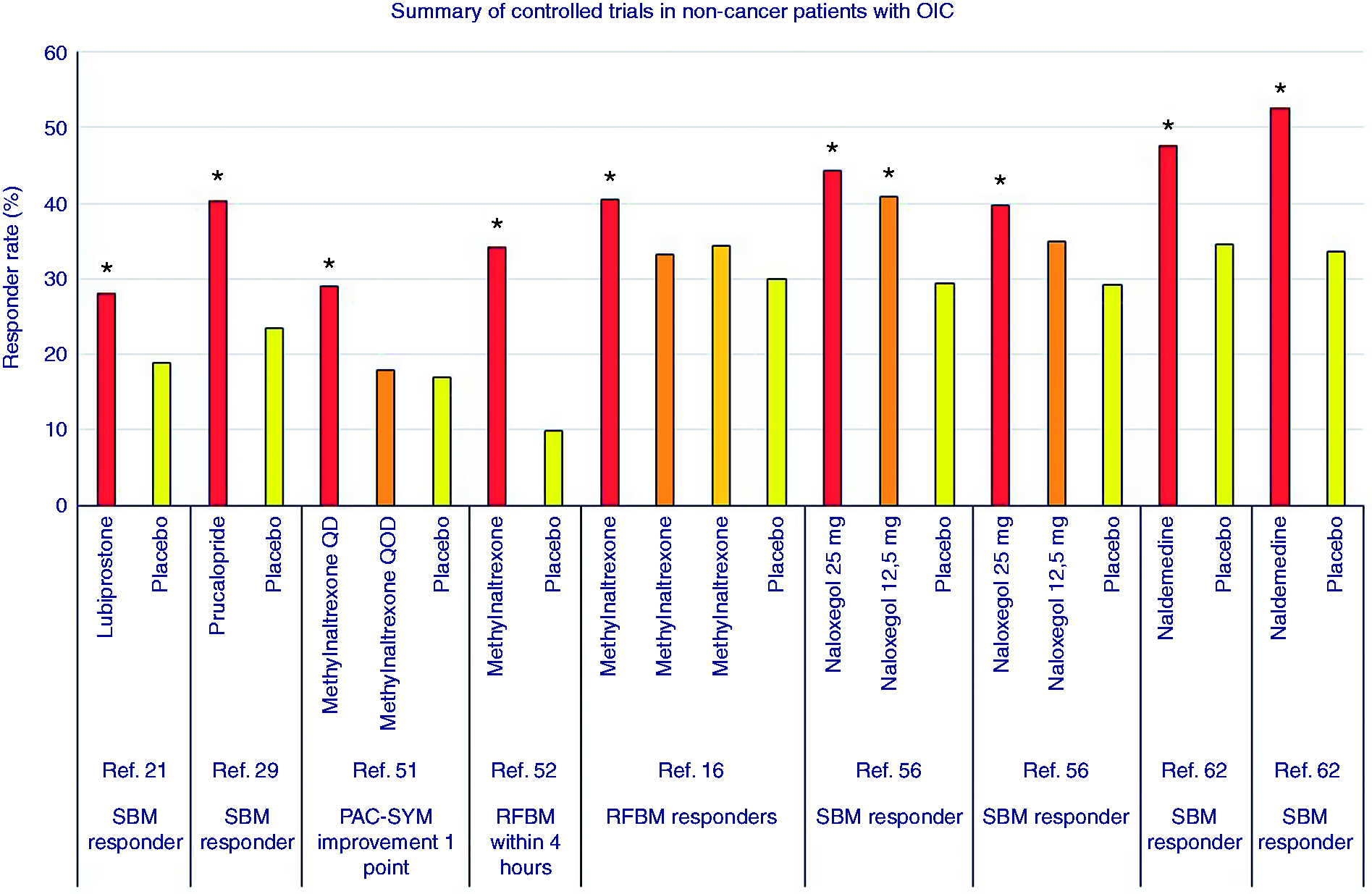
Multiple guidelines and reviews include use of polyethylene glycol (PEG), stimulant laxatives and stool softeners as first-line treatment in OIC.9,20 As they are inexpensive, widely available and considered generally safe, laxatives such as bisacodyl, sodium picosulphate, macrogol, and sennosides are considered first-choice treatment for OIC. 20 In addition, stimulant laxatives are traditionally used as rescue therapy in trials.17,21–23
Only a few studies have addressed laxative efficacy in OIC. In a randomised, double-blind, placebo-controlled trial performed in 57 patients enrolled in a methadone maintenance program, lactulose and PEG resulted in significantly fewer hard stools compared to placebo (p < 0.01). 24 A systematic review concluded that there were insufficient studies for statistical analysis and that there was a lack of evidence of significant differences between over-the-counter laxatives.25,26
Lubiprostone, an intestinal secretagogue
Lubiprostone, an orally administered, luminally acting chloride channel (CCl2) activator, has been studied in OIC treatment in adults with chronic, non-cancer pain. The US Food and Drug Administration (FDA) approved lubiprostone 24 µg twice daily for treatment of OIC in patients with non-cancer pain in the US and Canada. Through the mutual recognition procedure, it also gained approval in several European Union (EU) member states.
Its mode of action is the activation of the CCl2 channel, which induces chloride secretion into the lumen of the bowel. Sodium and water follow passively, thereby softening the stools. There is, however, no direct effect on the contractile apparatus.
Three randomised, double-blind, placebo-controlled, 12-week trials have been conducted. Jamal et al. found a significantly greater response rate for lubiprostone compared to placebo of 3.2 vs 2.4 mean change from baseline spontaneous bowel movements per week (SBMs), respectively (p = 0.001). This result reflected a number needed to treat (NNT) of 13. More patients in the lubiprostone group compared to the placebo group were overall SBM responders (27.1% vs 18.9%, respectively; p = 0.03). In addition, median time to the first SBM was significantly shorter after lubiprostone compared to placebo (23.5 vs 37.7 hours, respectively; p = 0.004). Finally, a significantly higher portion of patients who underwent lubiprostone treatment reported their first SBM within four, eight, 12 and 48 hours of the first dose (p ≤ 0.009). 13
In a second phase 3 trial enrolling 418 non-cancer pain patients with OIC, Cryer et al. found a significant change in SBM frequency/week favouring lubiprostone compared to placebo at week 8 (mean, 3.3 vs 2.4 SBMs/week, respectively; p = 0.004). At week 12, however, no significant difference was found. 23 A number of dropouts and fewer patients remaining in the study may have contributed to this difference. Furthermore, this study did not exclude patients taking methadone, and subsequent research showed a specific interaction with lubiprostone’s mode of action.
Methadone and morphine belong to two different classes of opioid receptor agonists: diphenylhepatanes and phenanthrenes, respectively. 27 It has been shown that methadone, but not morphine, inhibits Cl– secretion by the CCl2 channel in T84 cells, at baseline and after stimulation with lubiprostone. 27 Interference of methadone with either the CCl2 channel or the process required to transport Cl– may have contributed to the above-mentioned study not reaching statistical significance. 27 Finally, improvements favouring lubiprostone over placebo were seen for abdominal discomfort, straining, constipation severity and stool consistency. Therefore, Spierings et al. conducted a pooled analysis study from three randomised, double-blind, placebo-controlled, 12-week phase 3 trials.13,23,28 Pooled intention-to-treat analysis included 1272 patients (non-methadone, n = 1115; methadone, n = 157). A higher proportion of patients in the non-methadone group taking lubiprostone met the efficacy criteria compared to placebo (24.2% vs 16.9%, respectively; p = 0.002). This difference was not observed in the patients using methadone (11.9% vs 9.6% respectively; p = 0.692). Furthermore, subgroup analysis of patients who were on non-methadone opioids showed greater improvements for lubiprostone compared to placebo for mean change from baseline for constipation-associated symptoms such as bloating, straining, stool consistency, constipation severity and abdominal discomfort. 28
A long-term safety and efficacy open-label extension study by Spierings et al. showed a mean SBM frequency increase of 4.9 up to 5.3 per week, compared to 1.4 per week at baseline (p < 0.001). Thereby, 67.0%–84.1% of patients did not need rescue medication. 29 Lubiprostone was however not superior to senna, a contact-stimulant laxative, for OIC treatment. Bowel rescue medication was often needed in both groups, which could have confounded their results but reflects the complexity of the patient population. 30
The most common treatment-related AEs were nausea, diarrhoea and abdominal distension, with the majority mild or moderate in severity.23,29 In patient assessments of quality of life (PAC-QoL) and EQ-5D measures, no differences were found between lubiprostone and placebo. 13
Prucalopride
Prucalopride is a selective 5-hydroxytryptamine receptor 4 (5-HT4) agonist and enterokinetic agent with the most information and experience gained in chronic constipation (CC), and strong recommendation for its use in European guidelines. 31 Its mode of action, however, is upstream from and not directly targeting the mu-opioid receptor. 32
A multicentre, phase 2, randomised, double-blind, placebo-controlled trial has been performed in patients with chronic non-cancer pain who were on a stable dose of opioid agonists. 14 Apart from 10 mg bisacodyl and/or enemas as rescue medication, laxative use was not allowed. A total of 196 patients were randomised to placebo, prucalopride 2 mg or 4 mg. Over a four-week period, 40.3% compared to 23.4% of patients achieved an increase of ≥1 SBM per week from baseline for prucalopride 4 mg compared to placebo, respectively (p = 0.002). The primary outcome variable, the proportion of patients reaching ≥3 SMBs per week, was significant in favour of placebo only in the first week. Most frequent AEs of abdominal pain, nausea, diarrhoea and headache reported in OIC were similar to those reported in CC. No difference was seen in AEs between the different treatment groups, except for a slightly higher incidence of severe abdominal pain for 4 mg prucalopride.
A 12-week, randomised, double-blind, placebo-controlled trial to evaluate the efficacy and safety of prucalopride in individuals with chronic non-cancer pain suffering from OIC was started in 2010, but terminated early based on a non-safety–related business priority decision. A total of 169 patients were randomised to placebo and prucalopride 1 or 2 mg. The percentage of participants with an average frequency of ≥3 SBMs did not reach statistical significance (p = 0.305). 33
Linaclotide
Linaclotide is a 14-amino-acid peptide that, through binding with the guanylate cyclase-C receptor, increases cyclic guanosine monophosphate. This increase in cyclic guanosine monophosphate triggers a signalling-cascade that induces cystic fibrosis transmembrane conductance regulator activation. This leads to intestinal luminal release of chloride and bicarbonate, and passive sodium and water passage, accelerating intestinal transit. 34 Approval has been obtained for use in CC and constipation-predominant irritable bowel syndrome (IBS-C) in the US and for IBS-C in the EU, based on studies34–36 showing efficacy in these conditions. With clinical experience gained in these conditions, the drug is well known to gastroenterologists, and off-label use in OIC is not uncommon. Linaclotide does not directly address the motility effect of opioids, however, but may counteract anti-secretory effects. Most commonly, linaclotide caused GI-related AEs such as diarrhoea, flatulence, abdominal pain and nausea. Severe diarrhoea was found in 4.2% in the 600-µg group and resolved without intervention. 33 Contraindications for its use are known or suspected blockage. 37
A phase 2, randomised, double-blind, placebo-controlled trial of linaclotide administered to patients with OIC receiving opioids for non-cancer pain was completed in 2016, but no study results have been published. 38
Oxycodone hydrochloride and naloxone hydrochloride, an extended-release combination tablet
Naloxone, a peripherally acting relatively non-selective opioid antagonist, is available in a combination tablet with oxycodone, a frequently used analgesic opioid. The combination tablet was approved by the FDA and European Medicines Agency (EMA) to treat pain that is severe enough to require daily, around-the-clock, long-term opioid treatment, and for patients for whom alternative pain treatment options are inadequate.39–41 Oxycodone’s use has gained a firm position in non-cancerous pain treatment such as chronic back pain and osteoarthritis, and cancer pain. 42 Several randomised, placebo-controlled trials have shown superiority of oxycodone and naloxone in a prolonged release (PR) combination tablet formulation (OXN PR), compared to oxycodone PR (OXY PR) alone in maintaining bowel function (quantified as the bowel function index (BFI)) with equal analgesic efficacy and comparable safety.43–46 The 2:1 oxycodone/naloxone ratio has been found most suitable. 46
Naloxone undergoes first-pass metabolism for the most part (>97%) in the liver, which allows it upon slow release to exert an effect only on the peripheral GI receptors. 47 Despite low bioavailability there seems to be a maximal dose that can be given without reversing analgesic effects, which is variable amongst patients depending on their physical tolerance or need for high doses of opioids. 48 OXN PR reduced patients’ BFI, improved SBMs, and clinically relevant improvement in patient assessment of constipation symptoms (PAC-SYM) and PAC-QoL was observed.44,45,49,50 Most frequently reported AEs were constipation, nausea, headache, vomiting and diarrhoea. 48 Of all patients, approximately 14% withdrew because of AEs and 12% withdrew because of lack of efficacy. 44
Reports of loss of analgesic control and withdrawal, even if rarely present, hamper enthusiasm to use this combination preparation. This was mainly seen with rapid-dose up-titration.51,52 For safety and efficacy reasons, it is important that tablets be swallowed as a whole. Breaking or crushing them can cause rapid release of oxycodone, leading to faster absorption and even fatality. 41 Furthermore, the flexibility of adjusting analgesic dose and agent is limited as the combination is available only with oxycodone.
Peripherally acting mu-opioid receptor antagonists (PAMORA)
Subcutaneous methylnaltrexone
The US FDA and EMA approved methylnaltrexone, a quaternary ammonium derivative of naltrexone, in 2008 as a subcutaneous injection. It was the first mu-opioid receptor antagonist approved for the treatment of OIC in patients with advanced illness who responded insufficiently to laxatives. 53 In 2015, the EMA approval was extended to OIC treatment in non-cancer patients. Recommended doses are 8 mg for patients of 38–62 kg, and 12 mg for patients weighing 62–114 kg. Thereafter, dosing is performed at 0.15 mg/kg. The N-methyl group restricts the ability to cross the blood-brain barrier because of polarity and low lipid solubility, which prevents central pain reduction. 15
A four-week, randomised, placebo-controlled trial by Iyer et al. with methylnaltrexone, once daily (QD) or every other day (QOD), was compared to placebo in non-cancer patients with OIC. A difference in adjusted change (mean (SD)) vs placebo was seen for rectal symptoms (–0.26 (–0.42, –0.11); p = 0.001), stool symptoms (–0.33 (–0.51, –0.14); p < 0.001) and global scores (–0.25 (–0.39, –0.10), p < 0.001) in the PAC-SYM questionnaire for QD methylnaltrexone. In the QOD group an improvement was seen in stool symptoms (–0.26 (–0.45, –0.07); p = 0.008) and global score (–0.15 (–0.29, –0.01); p = 0.04). Abdominal symptoms and pain scores were not significantly different in either group compared to placebo. Fifty-three per cent of QD patients compared to 52% QOD patients and 41% of the placebo group met minimal improvement criteria. In addition, 29% of the patients in the methylnaltrexone QD group met the criteria for moderate clinical improvement compared to 18% for methylnaltrexone QOD and 17% for placebo. 15
Michna et al. conducted a randomised, controlled trial at 91 centres in the US and Canada in patients with chronic non-cancer pain and OIC. In total 469 patients were randomly assigned to subcutaneous methylnaltrexone 12 mg QD, 12 mg QOD, or placebo for four weeks, with discontinuation of all laxatives at study entry. Of patients who received methylnaltrexone, 34.2% achieved a rescue-free bowel movement (RFBM) within four hours of the first dose compared to 9.9% for placebo (p < 0.001). A significant difference in mean change of RFBMs per week from baseline was seen for QD and QOD methylnaltrexone of 3.1 (p < 0.001) and 2.1 (p = 0.01), respectively, compared to 1.5 for placebo. An NNT of five and 14, for QOD and QOD dosing, respectively, was needed to achieve ≥3 RFBMs per week. Most commonly reported AEs were abdominal pain, diarrhoea and nausea. In addition, hyperhidrosis was more prevalent for methylnaltrexone. 16
Most research has been conducted in OIC in advanced illness. During the first 18 months after approval, seven cases of bowel perforation, after administration of subcutaneous methylnaltrexone for OIC, were reported in the FDA Adverse Event Reporting System. 54 Although all patients had serious underlying conditions and were taking concomitant medication, the causal relationship of methylnaltrexone to the events could not be ruled out nor established. 55 Therefore, methylnaltrexone should be restrained in patients who are at risk for GI perforation such as peptic ulcer syndrome, Ogilvie’s disease, diverticular disease or in case of infiltrating malignancy. 56 High costs and mode of administration are limitations for clinical use of subcutaneous methylnaltrexone.
Oral methylnaltrexone
An oral formulation of methylnaltrexone bromide was recently evaluated for use in the treatment of OIC in adults with chronic non-cancer pain. Owing to often long-term chronic pain management and the ease of oral treatment compared to subcutaneous injection, the FDA approved its use.
A randomised, placebo-controlled, double-blind, phase 3 trial by Rauck et al. allocated 804 patients to 150, 300 or 450 mg or placebo QD for four weeks, followed by as-needed dosing for eight weeks. During this period, patients continued on their initial treatment before randomisation. Laxative therapy was discontinued, but rescue laxative treatment was allowed. The primary efficacy endpoint of the study was the mean percentage of days that resulted in an RFBM within four hours of dosing during weeks 1 to 4. Key secondary efficacy endpoints included the percentage of responders (≥3 RFBMs per week, with an increase of ≥1 RFBM per week from baseline for at least three out of four weeks) during weeks 1 to 4, and change in weekly number of RFBMs from baseline during weeks 1 to 4. Analysis showed comparable results for the 450 mg tablet compared to subcutaneous methylnaltrexone. 17 At the end of the QD period, a mean of 24.3% and 26.2% of the doses resulted in an RFBM within four hours, for the 300 mg and 450 mg tablet, respectively, compared to 19.2% for placebo (both p ≤ 0.05). The results of the dosing as-needed period were comparable (300 mg, 27.6%; 450 mg, 30.5%; placebo, 18.5%; p ≤ 0.01 for each). The most common AEs in the 450 mg group, all mild to moderate in intensity, were abdominal pain, nausea and diarrhoea. Similar to subcutaneous methylnaltrexone, the oral tablet should not be used in patients who are at risk for GI perforation. Finally, opioid analgesia at baseline compared to weeks 4 and 12 did not show a statistically significant difference. 17
Naloxegol
Naloxegol is a pegylated derivative of the mu-opioid receptor antagonist naloxone. Pegylation induces P-glycoprotein transporter-substrate properties, prevents passage of the blood-brain barrier and thus limits the potential for naloxegol to interfere with the central mediated pain relief. 18
Two randomised, placebo-controlled, double-blind, parallel-group, multicentre, phase 3 trials have been conducted. KODIAC 4 and 5 investigated the response rate (≥3 SBMs per week and ≥1 SBM over baseline for ≥9 of 12 treatment weeks and ≥3 of the final four treatment weeks) with naloxegol 12.5 or 25 mg QD compared to placebo. Naloxegol in a dose of 25 mg resulted in a significantly higher response rate compared to placebo in both trials (p = 0.001 and p = 0.02) but not for the 12.5 mg dose (p = 0.02 and p = 0.2, respectively). The response-rate differences between active treatment and placebo were 13.8% (95% confidence interval (CI), 1.6–26.0) with 12.5 mg and 19.9% (95% CI, 7.7–32.1) with 25 mg. In both studies, greater improvements were seen with 25 mg for straining, stool consistency and frequency of days with complete SBMs compared to placebo. The proportion of patients requiring rescue treatment with bisacodyl were similar in all treatment arms in both studies, and there was no interaction between baseline opioid dose and naloxegol regarding response rate. 18
Data pooling of patients entered in KODIAC 4 and 5 and categorised as laxative inadequate response (LIR) (any laxatives for ≥4 days within two weeks and continued stool symptom ratings of moderate, severe, or very severe) or further categorised when taking ≥2 laxative classes (LIR-2), showed consistent results compared to the earlier studies. 18 Efficacy analysis showed higher response rates in the pooled LIR population for 25 mg (95% CI 1.253–2.001; p < 0.001) and 12 mg (95% CI 1.106–1.797; p = 0.005) dosing compared to placebo with similar results for the LIR-2 subpopulation (naloxegol 25 mg: 95% CI 1.010–2.213, p = 0.04; naloxegol 12.5 mg: 95% CI 0.996–2.183, p = 0.05). Furthermore, median (95% CI) time to first SBM post-administration was 7.6 (5.2–18.9) and 19.2 (9.3–21.8) hours for 25 and 12.5 mg, respectively, compared to 41.1hours (30.9–47.7) for placebo. 57
KODIAC 8, a 52-week, multicentre, open-label, randomised, parallel-group, phase 3 study was conducted to evaluate long-term safety and tolerability of naloxegol in patients with pain and OIC. 58 Naloxegol was found to be generally safe and well tolerated with abdominal pain, diarrhoea and nausea as most prevalent AEs. These were mostly mild and moderate in intensity, occurred in the first 12 weeks, and resolved during treatment or after discontinuation. 58 These data are consistent with the data from the earlier phase 3 trials KODIAC 4 and 5.
Alvimopan
Alvimopan is a mu-opioid receptor blocker which was initially approved for treatment of post-operative ileus. More recently, four trials have been performed in OIBD22,59–61 with dose regimens of 0.5 mg, 1 mg,22,59,60 and 2 mg. 61 Two replicate, randomised, placebo-controlled, phase 3 trials were conducted. One found a statistically significant dose-related result of alvimopan 0.5 mg twice a day (BID) (3.51 vs 2.01 SBMs frequency increase per week; p < 0.001) for the primary outcome of ≥3 SBMs per week or ≥1 SBM increase from baseline; 22 however, this result was not confirmed by the other study. 59 This was due to an unexpectedly high placebo response (56%) and reduced efficacy of alvimopan. After FDA review, continued clinical development of alvimopan was permitted but the sponsor decided to discontinue.
Other drugs in development or under evaluation
Naldemedine
Naldemedine is the newest orally available, peripherally selective mu-opioid receptor blocker antagonist that is FDA approved for OIC treatment in adult patients. Two randomised, controlled, phase 3 trials have been conducted on the efficacy and safety of the drug in OIC patients with chronic non-cancer pain. 19 The primary endpoint of the study was the proportion of responders, defined as having ≥9 positive response weeks in the 12-week treatment period and ≥3 in the last four weeks of the 12-week treatment period. In the COMPOSE I and II trial, 47.6% compared to 34.6% (p = 0.002), and 52.5% compared to 33.6% (p < 0.0001) of participants in the naldemedine vs placebo group, respectively, were found to have ≥3 SBMs per week and an increase of ≥1 SBM per week. In addition, from baseline compared to the last two weeks, an improvement in least square mean difference (95% CI) of 1.3 (0.77, 1.83) and 1.40 (0.92, 1.88) in SBM frequency was seen for naldemedine vs placebo (p < 0.0001 for both values), in the COMPOSE I and II trial, respectively. This clinically significant effect was already present during the first week after treatment. GI-related AEs such as diarrhoea, nausea and abdominal pain were more prevalent in the naldemedine group but were mild to moderate in nature. Finally, there were no opioid withdrawal symptoms or interference with opioids’ analgesic efficacy. 19
The long-term safety of naldemedine was evaluated in patients with OIC and chronic non-cancer pain in a 52-week, randomised, double-blind, phase 3 study. Eligible patients had chronic non-cancer pain for ≥3 months, were receiving stable daily doses of opioids (≥30 mg oral morphine equivalents) for ≥1 month prior to screening, and had OIC. The primary endpoint was summary measures of treatment-emergent AEs. Secondary efficacy endpoints included change from baseline in the frequency of bowel movements on weeks 12, 24, 36, and 52. A total of 1246 patients were equally randomised to receive naldemedine or placebo. The proportion of patients that experienced treatment-related AEs was similar across both treatment groups, with diarrhoea being the most prevalent AE in the naldemedine group. In addition, other GI-related AEs such as abdominal pain or vomiting were more prevalent in the naldemedine treatment group compared to the placebo group, respectively. Similarly to the COMPOSE I and II trials, the severity of AEs was mild to moderate. In total eight deaths (naldemedine, n = 4; placebo, n = 4) occurred during this 52-week long-term safety study, but none were considered by the investigators to be treatment related. 21
Bevenopran
Bevenopran (CB-5945) is a PAMORA that was in development for the treatment of OIC. Two phase 3 clinical trials and one safety study were terminated prematurely in 2014 because of difficulties with enrolment. 62
Axelopran
Axelopran (TD-1211) is a once-daily, oral PAMORA on which four phase 2 trials have been completed. Four phase 2 trials have been completed in more than 400 patients with OIC. Up to now, data have been published in only one abstract. In this study, three oral doses of axelopran were evaluated in a five-week, double-blind, placebo controlled, and parallel-group study conducted in 217 chronic non-cancer pain OIC patients. At week 5, the mean change from baseline in weekly complete SBMs for axelopran patients with <5 years of OIC ranged from 1.7 to 2.3 vs 0.7, and 1.2 to 3.3 vs 0.6, respectively, for axelopran vs placebo with OIC ≥5 years. The most prevalent AEs were GI related, were associated with treatment initiation, and were mild to moderate in nature. 63 There has been no registration of axelopran starting phase 3 trials to date.
Conclusion and future directions
Chronic pain is a prevalent condition associated with large-scale chronic use of opioids leading to significant side effects such as OIBD and OIC. In addition, the general increase in use of opioids for pain has had an influence on the prevalence of these conditions. Because of high impact on QoL in these patients, adequate treatment is required. During early treatment, over-the-counter laxatives have shown to be justified. Prophylactic laxative use in patients taking opioids should be standard care, which will reduce the prevalence and burden of OIC. Several new medications have been identified, and their efficacy has been shown in qualitative trials. Thereby, new pharmacological treatments are emerging at a rapid rate. PAMORAs are the rational treatment choice in this condition when conventional laxative treatment does not improve patients’ symptoms. Finally, PAMORAs are becoming more practical as treatment options are emerging in tablet form compared to earlier subcutaneous injections.
Footnotes
Declaration of conflicting interests
J.P. has nothing to declare. T.V. has served on the speakers bureau of Kyowa Kirin and Abbott, and has consulted for Shire. J.T. has served on the advisory board of Kyowa Kirin and Shionogi, and on the speakers bureau of Kyowa Kirin.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Informed consent
Not applicable as this paper is a review and does not involve subjects or patients.
Ethics approval
Not applicable as this paper is a review and does not involve subjects or patients.