
Abstract
Background
Vedolizumab is a recently available monoclonal antibody targeting α4β7 integrin for the treatment of ulcerative colitis (UC) and Crohn’s disease (CD).
Objective
The objective of this article is to evaluate the efficacy of vedolizumab induction therapy in anti-TNF-refractory/intolerant UC and CD patients in real life.
Methods
A cohort of 149 moderately to severely active UC and CD patients who failed or showed intolerance to at least two TNF antagonists participated in a medical need program and received vedolizumab in 37 Belgian centers (April–September 2015). Rates of clinical response and remission were retrospectively evaluated at Week 10 for UC and Week 14 for CD using the physician’s global assessment (PGA), Mayo score and Harvey Bradshaw index (HBI) or Crohn's disease activity score (CDAI) scores.
Results
Eighty-four patients (29 UC, 55 CD) had sufficient data for analysis. For UC patients, clinical response was observed in 76% based on PGA and 59% based on the Mayo score. The corresponding percentages for CD patients were 80% for PGA and 65% for HBI/CDAI. Clinical remission rates were 10% and 40% for UC and CD, respectively. Steroid-free remission was observed in respectively 10% and 35%. Globally, corticosteroids were stopped in 14 out of 48 patients (29%). No new safety signals were reported.
Conclusion
Up to 70% TNF-refractory/intolerant UC and CD patients achieved a clinical response after 10 to 14 weeks of vedolizumab treatment in this real-life cohort.
Key summary
Vedolizumab is effective in patients with inflammatory bowel disease refractory/intolerant to at least two tumor necrosis factor antagonists. In real life clinical response after induction therapy is observed in about 70% of patients. Steroid-free remission is found in 10% of ulcerative colitis patients and 35% of Crohn’s disease patients.
Introduction
Inflammatory bowel disease (IBD) is a chronic, relapsing condition that involves uncontrolled systemic inflammation of the gastrointestinal tract. Ulcerative colitis (UC) and Crohn’s disease (CD) are the two most common forms of IBD. Although the exact cause of UC and CD remains largely unknown, considerable progress has been made in the understanding of their underlying immunological basis. This has led to the development of biological agents that specifically target key mediators and biological pathways involved in the pathogenesis, such as tumor necrosis factor α (TNFα) antagonists. Despite the fact that TNF antagonists are highly effective, a significant proportion of patients face suboptimal outcomes due to primary or secondary therapy failure, or due to side effects leading to discontinuation of the drug. 1
Leukocyte trafficking is a new therapeutic target in the treatment of UC and CD, with vedolizumab as the current lead compound.2,3 The inflammation in UC and CD is characterized by a massive infiltration of leukocytes into the intestinal mucosa. This multistep process is tightly regulated through complex interactions between circulating leukocytes and intestinal vascular endothelial cells. Adhesion of the infiltrating leukocytes to the endothelium is mediated by binding of integrin molecules expressed on the leukocyte surface to cellular adhesion molecules on the surface of the intestinal endothelial cell. Vedolizumab is a humanized, gut-selective monoclonal immunoglobulin (Ig)G1 antibody directed against the α4β7 integrin heterodimer. It blocks the interaction of α4β7 integrin with mucosal addressin cell adhesion molecule-1 (MAdCAM-1) on intestinal vasculature, thereby preventing leukocyte binding to the endothelial surface and extravasation into affected tissue. 4
Clinical studies including more than 2800 patients with active moderate to severe UC or CD demonstrated that vedolizumab is effective as induction and maintenance therapy with a favorable safety profile. Incidence rates of serious or opportunistic infections were low and no increased risk for malignancies was observed.5–8 Although the therapeutic benefits and risks of vedolizumab treatment have been evaluated in several Phase II and III clinical studies, beneficial effects beyond the clinical trial population are not necessarily guaranteed. 9 Predominantly in patients who previously failed at least one TNF antagonist, efficacy of vedolizumab remains unclear with a later and worse response than in anti-TNF-naïve patients. 7 This retrospective study describes the efficacy of vedolizumab as third-line therapy in real-life circumstances in patients refractory to at least two TNF antagonists.
Materials and methods
Patients
A medical need program (MNP) was set up by Takeda Belgium to provide vedolizumab to patients with moderate to severe IBD and known primary or secondary failure or intolerance to at least two TNF antagonists before reimbursement. Data were collected retrospectively from 149 patients who were enrolled in this MNP and were treated with vedolizumab between April and September 2015 in 37 centers in Belgium. The MNP protocol was developed by Takeda and approved by the ethics committee of the University Hospital of Leuven. The study protocol for the retrospective analysis of the MNP patient data was developed by one author (MDV) independently from Takeda and approved by the ethics committee of the University Hospital of Ghent (EC UZG 2015/1144-date approval November 16, 2015). The study protocol conforms to the ethical guidelines of the 1975 Declaration of Helsinki as reflected in a prior approval by the institution’s human research committee. All patients provided written informed consent. Patients’ age, age at diagnosis, disease duration, disease extent (Montreal classification), 10 disease activity scores, presence of fistulae, surgical history, previous and concomitant IBD-related medication such as 5-aminosalicylic acid (ASA) derivatives (mesalazine or sulfasalazine), steroids (budesonide, beclomethasone, methylprednisolone or prednisolone), immunomodulators (azathioprine, 6–mercaptopurine, methotrexate, cyclosporin) and TNF antagonists (infliximab, adalimumab, golimumab, certolizumab pegol), and baseline C-reactive protein (CRP) levels were recorded into a database for analysis.
Treatment schedule
Vedolizumab was administered according to the label, with intravenous infusions of 300 mg at Week 0, 2 and 6 (induction phase), with an optional infusion at Week 10 for CD patients. The decision to administer an additional vedolizumab dose at Week 10 in the group of CD patients was based on studies showing delayed responses in CD compared to UC and is incorporated into the prescribing information for vedolizumab.5–7 In case of clinical response, therapy was continued with 300 mg intravenous infusions every eight weeks (maintenance phase).
Efficacy evaluations
Clinical response and clinical remission were assessed at Week 10 for UC patients and at Week 14 for CD patients. Evaluation of the clinical response was based on the physician’s global assessment (PGA) and measurement of the following disease activity scores: total Mayo score11,12 for UC patients, and Crohn’s disease activity index (CDAI) 13 or Harvey-Bradshaw index (HBI) 14 for CD patients. PGA was made in all patients. Evaluation of clinical response, defined as a reduction in the total Mayo score of at least three points and a decrease from baseline of at least 30% for UC patients, and a reduction in the CDAI score of at least 100 points or a reduction in the HBI score of at least three points for CD patients, was possible only when data were available both at baseline and at Weeks 10/14. Remission was defined as a total Mayo score of ≤2 for UC patients, and a CDAI score of ≤150 or an HBI score ≤4 for CD patients. These data were available for all included patients.
Safety
Adverse effects were reported by the treating physician.
Statistical analysis
Statistical analyses were performed with SPSS 23 (IBM, Armonk, NY, USA) software. Descriptive statistics (medians and corresponding interquartile range (IQR)) were reported whenever applicable.
Results
Patient disposition and baseline demographics
Baseline demographics and disease characteristics.
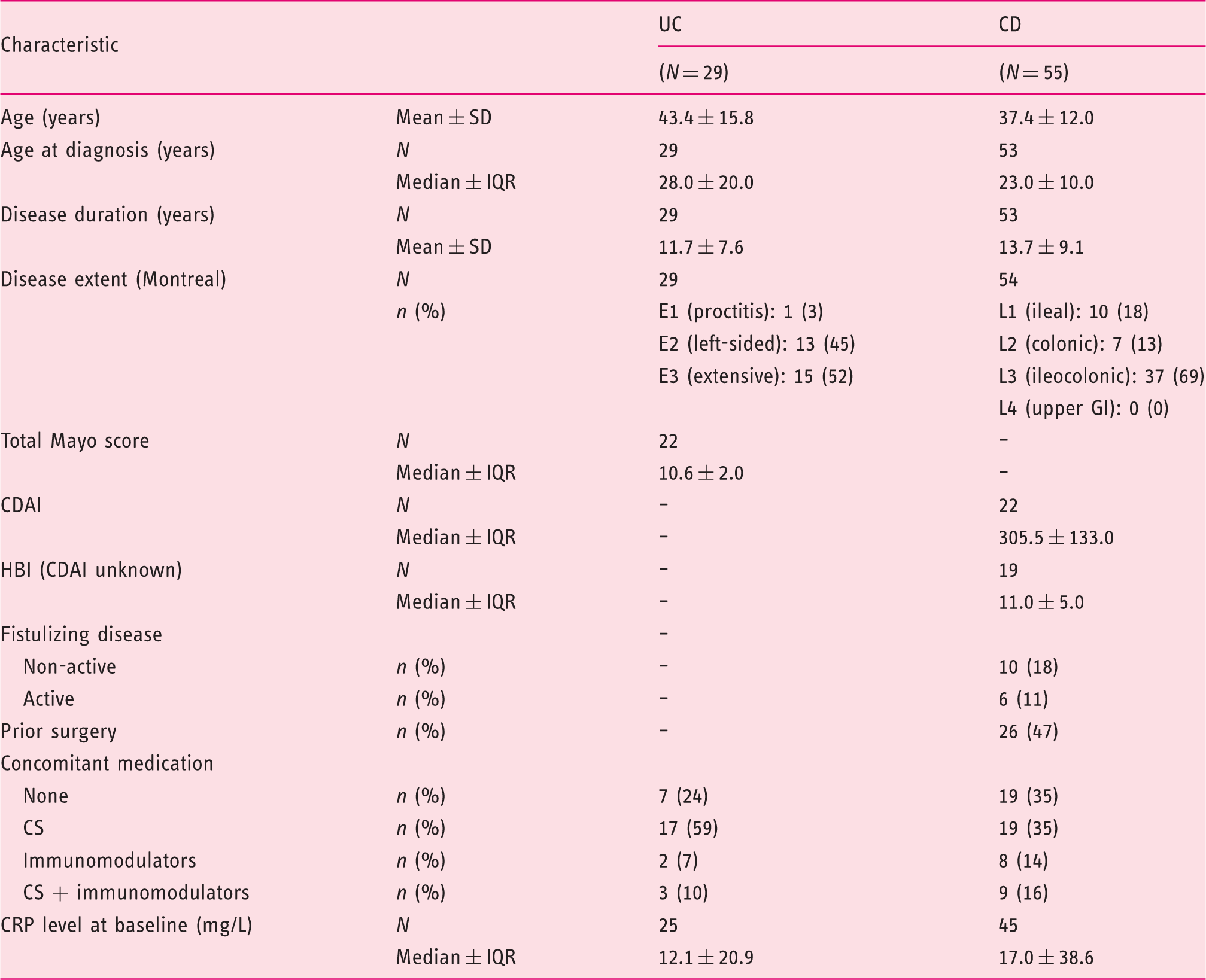
N: number of participants with data; n: number of participants with this observation; SD: standard deviation; IQR: interquartile range.
UC: ulcerative colitis; CD: Crohn’s disease; CDAI: Crohn's disease activity score; HBI: Harvey Bradshaw index; CS: corticosteroids.
UC
The median (±IQR) total Mayo score of 10.6 (±2.0) at baseline reflected the severity of the disease. Moreover 20 out of 29 patients were on corticosteroids at baseline.
CD
The median (± IQR) disease activity score of 305.5 (± 133.0) for CDAI or 11 (± 5) for HBI illustrated the severity of the disease. Twenty-eight out of 55 patients were on corticosteroids at baseline. Sixteen out of 55 patients had additional fistulizing disease (with active drainage in 10 patients). Forty seven percent of patients had previous surgery.
Efficacy evaluations
UC
At Week 10, a clinical response was observed in 76% of UC patients according to PGA. Clinical response based on the predefined reduction in Mayo score was observed in 59% of UC patients (data available from only 22 patients). Clinical remission was seen in 10% of patients and none of these patients required oral corticosteroids at that time (Figure 1). Three of 20 patients taking steroids at baseline were able to discontinue this therapy by Week 10 (of whom one patient was in clinical remission).
Efficacy of vedolizumab induction therapy in ulcerative colitis (UC) (week 10) and Crohn's disease (CD) (week 14).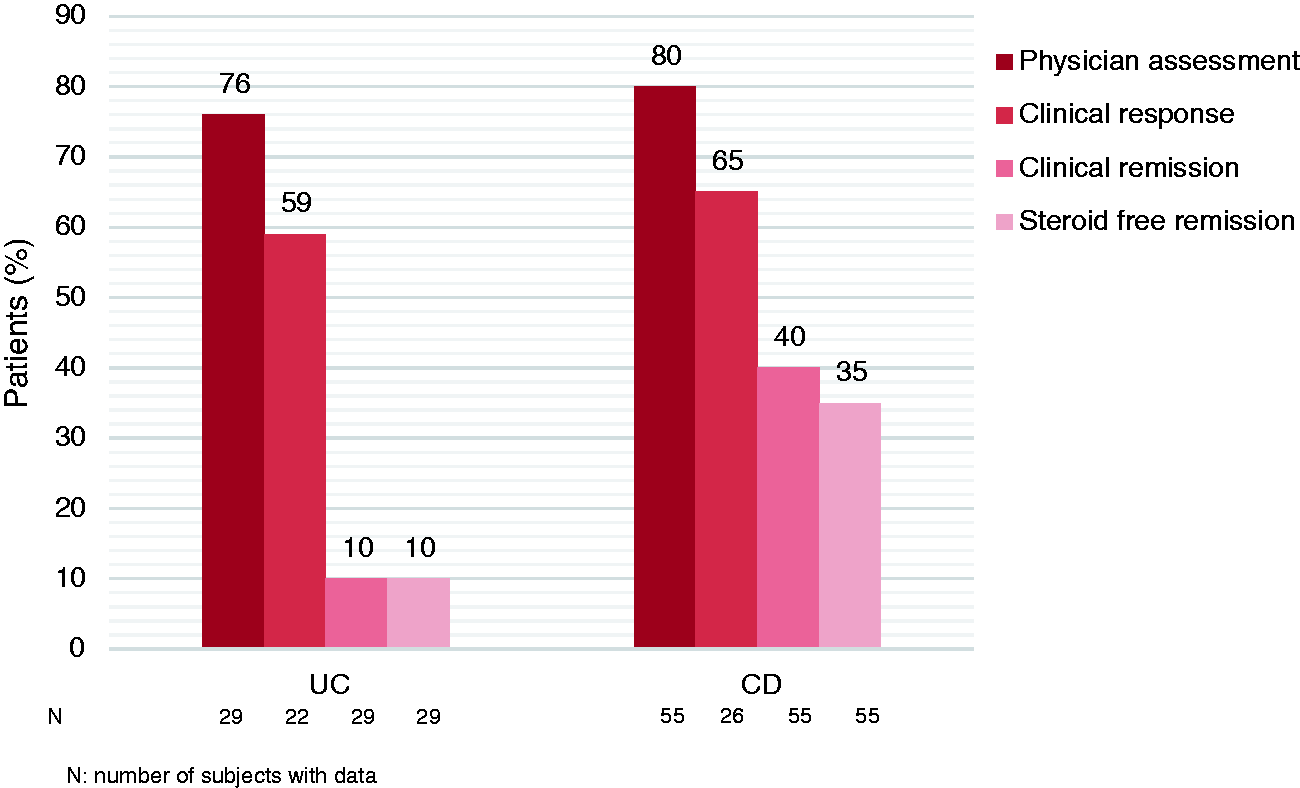
CD
At Week 14, a clinical response was seen in 80% of CD patients based on PGA. Clinical response based on the predefined reduction in CDAI or HBI score was observed in 65% of CD patients (data available for only 26 patients). Clinical remission was observed in 40% of CD patients, and the majority of those patients were steroid free (19 out of 22 patients) (Figure 1). Eleven of 28 patients taking steroids at Week 0 were able to discontinue this therapy by Week 14 (of whom seven patients were in clinical remission).
Safety
No deaths or serious adverse events were reported. Adverse events reported during treatment were upper respiratory tract infection, skin eruption, chills during first infusion, headache and cluster headache; only headache was reported by more than one patient (n = 3). Eight patients needed colectomy because of non-response (four UC and two CD patients), acute subobstruction (one CD patient) or perforation during colonoscopy (one CD patient).
Discussion
In this real-life multicenter Belgian study, the efficacy and safety of vedolizumab as induction therapy were evaluated in UC and CD patients with a documented failure/intolerance to two or more TNF antagonists. This study confirmed that vedolizumab is effective in this subgroup of very refractory patients, as illustrated by the high clinical response rates (∼70%) achieved at Week 10 (UC) and 14 (CD). The clinical response rates based on PGAs (76% and 80% for UC and CD, respectively) were in line with the response rates based on the change in disease activity scores (59% and 65%). In general, the observed clinical responses based on disease activity scores in our study were similar or higher than rates reported in other real-life studies after 14 weeks of therapy, ranging from 48.9% to 64% in CD patients, and from 53.9% to 57.4% in UC patients.15–17 Differences in patient population, sample size and use of different measurements to assess clinical improvement can explain differences in magnitude of effectiveness across studies. The French CD population studied by Amiot et al., with a similar median disease duration (12 years) and TNF antagonist experience (91% failed in one or more previous TNF antagonist therapy), demonstrated similar response and remission rates in CD patients (64% and 36%, respectively). 16
The clinical remission rate in our CD cohort (based on CDAI or HBI scores) was 40% at Week 14, which is higher than the one reported at Week 6 in the large pivotal GEMINI II trial (31.4%). 6 This can be explained by the earlier timepoint of measurement in the GEMINI II trial (Week 6 versus Week 14 in our study), whereas it has been shown that the therapeutic effect of vedolizumab in this refractory patient group becomes clear only beyond Week 10. 7 The lagging in vedolizumab kinetics was shown in other real-life studies in TNF antagonist-experienced CD patients in whom clinical effectiveness continued to improve from Week 6 to Week 14.15,18 Results reported by Dulai et al. even demonstrated greatest benefit after 25 weeks of therapy in terms of clinical remission. 19
The analysis of the GEMINI trials suggested that vedolizumab could be more beneficial in UC than in CD patients. However, in the present study, comparable response rates were observed in UC and CD patients (65% in CD at Week 14, 59% in UC at Week 10). The results obtained in a United States prospective study (48.9% in CD, 53.5% in UC), 17 a German study (60.8% in CD, 57.4% in UC) 15 and a French study (64% in CD, 57% in UC) 16 show no significant differences in response rates in UC and CD patients and therefore confirm our findings.
Remarkably, remission rates in UC patients (10%) were lower compared to the ones previously reported in real-life studies (ranging from 23.5% to 55%).15–18 This difference cannot be attributed to age, disease duration or extent of involvement since data for these parameters were similar among the different studies. However, the activity scores used to define remission in UC varied among the different studies. In the present study, a reduction in the total Mayo score to a maximum of two points was used to define clinical remission (including endoscopic score). In the study by Shelton et al., clinical remission was defined as a simple clinical colitis activity index (SCCAI) ≤2 or based on PGA. 17 Baumgart et al. used a partial Mayo score of ≤1, whereas Vivio et al. used a partial Mayo score of ≤2 to define remission.15,18 This makes interpretation and comparison of results across different studies very difficult. Moreover, in the present study the efficacy of vedolizumab for treatment of UC was evaluated at Week 10, while this was performed at Week 14 in the other real-life studies. Compared to placebo-controlled clinical trial (GEMINI trial) and real-life data,16,17 the median total Mayo score at baseline was slightly higher in the present study reflecting a more severe disease at baseline in our study. More important, all UC patients experienced failure due to resistance or intolerance to two or more TNF antagonists, while less than half of the patients in the GEMINI trial had previous experience with TNF antagonists. Similarly, in the other real-life cohorts only 50% and 71% of patients failed anti-TNF.15–17 Remission rates in those studies varied between 24% and 39%. However, no data are provided about rates in those severe refractory subgroups, although we know from the GEMINI trials that vedolizumab in anti-TNF-naïve patients is clearly less effective in anti-TNF failures.
Steroid use in our population (57%) is in agreement with those observed in different studies, varying between 29% and 84%.15–20 We reported a steroid-free remission in 10% of UC patients similar to those reported in the literature15–17 but lower than reported in the Swedish National registry (33%). 20 However, in this cohort high remission rates can be at least partly explained by low clinical disease activity at inclusion.
In CD patients steroid-free remission was observed in 35% of patients, similar to results by Amiot et al. (31%) 16 and Eriksson et al. (40%) 20 but higher than those reported by Baumgart et al. (20%) 15 and Shelton et al. (23%). 17
In the present study, with a short follow-up no specific safety concerns were noted, which conforms to the integrated vedolizumab safety analysis performed by Colombel et al., showing low incidence rates of serious infections, infusion-related reactions and malignancies over an extended treatment period with more than 4800 patient-years. 8
Limitations of this study include the open-label design, the limited number of patients compared to large randomized trials and the retrospective data collection leading to missing data and short follow-up period. Data from the PGA and remission rates were available for all 84 patients included in the analysis, while disease activity scores at Week 0 were missing in a significant number of patients, mainly in the CD group, in whom a pre-treatment score was available for less than half of the patients. Caution is thus required when interpreting response data, although they are in line with results from other real-life studies. Because of the lack of baseline data, analysis of predictors of effectiveness was not possible in this study. It is expected that the general IBD patient population will show an even more favorable response than the described highly refractory patient cohort. Advantages of this study include the real-life design and multicenter collection of patients, which ensures heterogeneity in patient inclusion.
In summary, this real-life study performed in multiple Belgian IBD centers clearly showed that vedolizumab is an effective induction therapy in approximately 70% of UC and CD patients resistant/intolerant to at least two TNF antagonists.
Footnotes
Acknowledgment
Medical writing assistance was provided by Emtex BVBA (Sint-Gillis-Waas, Belgium) supported by Takeda Belgium.
Declaration of conflicting interests
MDV received consultancy fees from Takeda, Abbvie, Janssen and educational grants from Ferring and Abbvie. BD, TH, AE, MVG and EM have no conflict of interests; SV has received grant support from AbbVie, MSD, Pfizer and Takeda; received speaker fees from AbbVie, MSD, Takeda, Ferring, Dr. Falk Pharma, Hospira, Pfizer Inc and Tillots; and served as a consultant for AbbVie, MSD, Takeda, Ferring, Genentech/Roche, Shire, Pfizer Inc, Galapagos, Mundipharma, Hospira, Celgene, Second Genome, and Janssen; EL has received Research Grant fromTakeda. Educational Grant: MSD, Abbvie, Takeda. Speaker Fees: Abbvie, Ferring, MSD, Chiesi, Mitsubishi Pharma, Hospira, Janssen, Takeda. Advisory Board: Abbvie, Ferring, MSD, Takeda, Hospira, Mitsubishi Pharma, Celltrion, Prometheus, Celgene, Janssen; FM has received speakers fee from Takeda; PB has received consulting fees from Dr Falk Benelux, Hospira, Janssens-Cilag, MSD, Mundipharma, Roche, Pfizer and Takeda, has received lecture fees from Abbvie, Takeda and Vifor Pharma, and has received an educational grant from Abbvie; FB has received research grants from Abbvie, Chiesi, Ipsen, MSD, Roche. Speakers and consultancy fees from Abbvie, Falk, Ferring, Janssen, Mundipharma, MSD, Pfizer, Takeda, Vifor; CR has received consulting from fees, lecture fees and travel accomodations from AbbVie, Takeda, MSD, Mundipharma, Hospira, Ferring, Janssen; MF has received – Research grant from Takeda; Speakers fee: Abbvie, Boehringer-Ingelheim, Chiesi, Falk, Ferring, Janssen, Mitsubishi Tanabe, MSD, Takeda, Tillotts, Zeria; Consultancy: Abbvie, Boehringer-Ingelheim, Ferring, Janssen, MSD; PH has received consulting fees from Abbvie, Takeda and Janssen; speaker's fees from Ferring, Falk Pharma, Vifor Pharma, Tillotts Pharma, Chiesi, Takeda and Abbvie; OD received speaker fee or consultancy: Abbvie,Ferring, Janssen, MSD, Takeda; DF received educational grants from Abbvie, Takeda, MSD, and received honorarium fee for lectures or consultancy from Ferring, Falk, Chiesi, Abbvie, MSD, Centocor, Pfizer, Amgen, Janssen, Mundipharma and Hospira.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Ethics approval
The study protocol was approved by the ethics committee of the University Hospital of Ghent (EC UZG 2015/1144-date approval November 16, 2015).
Informed consent
All patients provided written informed consent.