
Abstract
The gut microbiota has recently been recognized as a major environmental factor in the pathophysiology of many human diseases. The anatomical and function connection existing between gut and liver provides the theoretical basis to assume the liver is a major target for gut microbes. In the last decades, numerous studies reported an altered composition of gut microbiota in patients with liver cirrhosis and a progressively marked dysbiosis with worsening of the liver disease. The risk of developing hepatocellular carcinoma, the deadliest complication of liver cirrhosis, is widely variable among cirrhotic patients, thus suggesting a complexity of genetic and environmental factors implicated in hepatocarcinogenesis. Gut microbiota is now emerging as a plausible candidate to explain this variability.
In this manuscript we review the human and the experimental evidence supporting the potential implication of gut microbiota in the promotion, progression and complication of liver disease.
Introduction
Hepatocellular carcinoma (HCC) is the fifth most common type of cancer and the third leading cause of cancer-related death globally. 1 HCC develops in the context of liver cirrhosis in the majority of cases and remains the leading cause of death among patients with advanced liver disease. 2 The risk of developing HCC is widely variable among cirrhotic patients, thus suggesting that, beyond cirrhosis itself, multiple additional genetic and environmental factors can be implicated in hepatocarcinogenesis. The gut microbiota has recently been recognized as a major environmental factor influencing the pathogenesis of several human diseases, 3 including liver cirrhosis4,5 and its complications. 6 The gut microbiome is a complex and dynamic microbial community, mostly constituted of bacteria, but also comprising fungi, protozoa, archea and viruses that inhabits the intestine. 7 In physiological conditions the intestinal microbiota has a symbiotic relationship with its human host to which it provides metabolic, trophic, immunological and defense functions. 7 The balanced interaction among bacteria, epithelium and gut immune system is a prerequisite to guarantee the paradoxical dual function of the human “healthy” gut that absorbs nutrients while limiting the access of pathogen bacteria and/or microbial-derived molecules (lipopolysaccharide (LPS), bacterial DNA, flagellyn, peptidoglycans, etc.) to the portal circulation and the liver. 8 The intestinal barrier, an anatomical-functional structure composed of an epithelial layer (mechanical barrier), mucus, immunoglobulin (Ig)A and antimicrobial products (secretory barrier) and the gut associated lymphoid tissue (immune barrier), regulates the trafficking throughout the intestinal wall. 9 A small quantity of selected microbes and their products physiologically enter the portal venous blood and reaches the liver where they interact with an enormous number of hepatic non-parenchymal cells that regulate the innate and adaptive immune response.10,11 In the liver, specific receptors such as Toll-like receptors (TLRs) and nucleotide binding oligomerization domain-like receptors (NLRs) recognize bacterial products, inducing the transcription of pro-inflammatory cytokines and chemokines by intracellular signaling cascades. 12 However, the constant low-level exposure to bacterial components renders the cells refractory to stimulation by TLR, a phenomenon known as “endotoxin tolerance,” leading also to active immune suppression via cytokines such as interleukin (IL)-10 or transforming growth factor beta (TGFβ), hepatocyte growth factor or hepatic stellate cells (HSC)-derived retinoic acid. 13
The integrity of the intestinal barrier is crucial to the anatomical and functional connection existing between gut and liver, summarized in the concept of the “gut-liver axis.” 14 Indeed, the liver receives approximately 70% of its blood supply from the intestine and delivers into the gut several substances beneficial to the intestinal trophism and functioning. 15 When quantitative and qualitative changes in gut microbiota (dysbiosis) occur, the integrity of the intestinal barrier is compromised with a consequent “leaky gut” and a pathological bacterial translocation (BT). 15 The microbiota-driven activation of Kupffer cells, the macrophages of the liver, leads to the release proinflammatory cytokines such as tumor necrosis factor alpha (TNFα), whereas the stimulation of HSC promotes the development and the progression of liver fibrosis mainly through TLR-4-dependent nuclear factor (NF)kB activation. 16 The inflammasome, the multiprotein complex that senses signals from pathogens and damaged cells, also contributes to the microbiota-driven liver injury and the hepatic immune response activation.17,18 The inflammasome activation peculiarly requires a double signal to induce the inflammatory response and leads to caspase-1 activation, which proteolytically activates the cytokines. This inflammatory response is amplified by IL-1β, which in turn provides positive feed-forward stimulation for pro-inflammatory molecules. 19
Clinical and experimental evidence convincingly supports the role of gut microbiota in many different stages of liver diseases. However, its contributory role in hepatocarcinogenesis is still an emerging issue. Thereby, in this manuscript we will focus on the human and the experimental evidence supporting this association.
Gut microbiome and liver cirrhosis
Liver cirrhosis, the end stage of different types of hepatic injury, is characterized by a derangement of hepatic architecture and portal hypertension. 20 The increase in portal pressure elicits structural modifications of the intestinal wall including vascular congestion, edema, fibromuscolar proliferation, thickening of the muscolaris mucosae and reduction or loosening of the tight junctions (TJ).21,22 As a consequence, intestinal permeability increases by both paracellular and transcellular movements, hesitating in a pathological BT. 23 On the other side, a low gastric acid secretion, an impaired intestinal motility, a reduced antimicrobial activity of defensins, decreased levels of mucosal IgA, and modifications in bile acids (BAs) secretion are reported in patients with cirrhosis. 24 Bile acids exert antimicrobial effects by directly damaging the membrane of bacterial cells and indirectly through the activation of antibacterial molecules through the farnesoid X receptor (FXR), a nuclear receptor for bile acids. 25 FXR also regulates the expression of genes crucial in preventing bacterial overgrowth and maintaining the integrity of the intestinal epithelium. 26
In summary, cirrhosis and portal hypertension directly impair intestinal permeability and indirectly affect the composition of gut microbiota, thus facilitating BT and worsening liver disease.27,28
Gut microbiota composition in patients with liver cirrhosis and HCC.

C: cirrhosis; CD: Crohn's Disease; HCC: hepatocellular carcinoma; HC: healthy controls; HBV: hepatitis B virus; HE: hepatic encephalopathy; DGGE: denaturing gradient gel electrophoresis; OGD: Other Gastrointestinal Disease; PCR: polymerase chain reaction; rRNA: ribosomal RNA.
Significant differences between controls and cirrhotics, but no difference between HE and non-HE patients.
Significant differences between controls, HE, and non-HE patients.
A reduced abundance of Bacteroidetes with an increased level of Proteobacteria and Fusobacteria at the phylum level, and an increased level of Enterobacteriaceae, Veillonelaceae and Streptococcaceae with a reduced level of Lachnospiraceae at the family level, was found in fecal samples of cirrhotic patients with respect to healthy controls. 29 Interestingly, the severity of the hepatic disease positively correlated with Streptococcaceae and negatively correlated with Lachnospiraceae levels. The Lachnospiraceae family includes such commensal genera as Coprococcus, Pseudobutyrivibrio and Roseburia, which benefit the host by producing short chain fatty acids (SCFAs) that exert a useful trophic, metabolic and immune-modulating effect. These modifications were independent of the etiology of liver disease, suggesting that cirrhosis itself can account for the altered composition of gut microbiota. 29 Similar results were reported by Bajaj et al., who described a progressively marked dysbiosis with the worsening of the liver disease while patients with a non-evolving disease course had a stable gut microbiota composition. 30 In details, the reduction of the autochthonous taxa such as Lachnospiraceae, Ruminococcaceae and Clostridiales XIV, and the relative increase of non-autochthonous taxa such as Staphylococcaceae, Enterococcaceae and Enterobacteriaceae, were linked to liver failure and endotoxin plasma levels. Interestingly, compared to non-alcoholic cirrhotic patients, alcoholic cirrhotics showed higher levels of Enterobcteriaceae and endotoxemia. The cirrhosis to dysbiosis ratio (CDR) is the ratio between autochthonous and non-autochthonous taxa, negatively correlated with endotoxemia and was highest in healthy controls, lower in compensated cirrhotic patients and lowest in decompensated cirrhotic patients. 30
Interestingly, Qin et al. found higher proportion of bacteria of buccal origin such as Streptococcus and Veillonella in the gut microbiome of cirrhotics, thus suggesting that the oral microbiota invades the gut and contributes to the progression of the disease in cirrhotics. 31 Indeed, the reduced gastric acid secretion and the modification in BAs secretion observed in cirrhosis may facilitate the intestinal colonization of bacteria of buccal origin. In contrast a fecal-like microbiota rather than an oral-like one, better correlated with end-stage liver disease, especially in patients with previous HE episodes, in whom an increase of potentially pathogenic families such as Enterobacteriaceae and Enterococcaceae and a decrease of autochthonous bacteria, was observed. 31
The mucosal microbiota, which resides inside the mucus layer adherent to the mucosa layer, is remarkably different and more stable over the time than the luminal counterpart. The microbiota adherent to mucosa layer constantly cross-talks with the host, therefore, the analysis of the fecal microbiota alone may be unrepresentative of the real composition of the gut microbiota and of its interplay with the host. In cirrhotic patients the stool microbiome differed from that of the corresponding colonic mucosa. Interestingly, the microbiota composition significantly differed between patients with and without HE only in mucosa but not in stool samples. Specifically, Veillonella, Megasphaera, Bifidobacterium, and Enterococcus genera, members of Firmicutes phylum, were prevalent in HE whereas Roseburia was more abundant in the non-HE group. 32
The alterations of the mucosal microbiota composition in liver cirrhosis are not limited to the lower digestive tract. Indeed, the analysis of duodenal mucosal microbiota in cirrhotic patients versus healthy controls showed remarkable differences with higher inter-individual variations. 33 At the genus level, a higher abundance of Atopobium, Dialister, Veillonella and Megasphera was observed in cirrhotics whereas Hemophilus, Neisseria and SR 1 genera incertae sedis were overrepresented in the control group.
Although clinical and experimental data sufficiently support the hypothesis that gut microbiota can be implicated in the pathogenesis of liver cirrhosis and its complications, we are still far from translating this information to the clinical setting. The limited number of patients analyzed, the non-stratification of patients according to liver disease etiology and, not least, the non-standardized methodology and the associative nature of the studies conducted so far, contribute to making the implication of gut microbes in the development or progression of liver diseases still speculative.
Gut microbiome and HCC: Animal models and human studies
In spite of the large amount of data on gut microbiome composition in cirrhosis, only little and recent evidence is available in liver cancer. A culture-based study reported higher levels of Escherichia coli in cirrhotic patients with HCC with respect to those without HCC, in spite of the etiology and severity of liver disease. 34 By a metagenomics approach, an altered microbiome profile was also revealed in the tongue coat of patients with HCC compared to healthy controls. 35 Overall 38 operational taxonomic units assigned to 23 different genera were able to distinguish HCC patients from matched healthy individuals. Strikingly, Oribacterium and Fusobacterium were microbial biomarkers of HCC. Interestingly, genes in the categories related to nickel/iron-transport, amino acid-transport, energy-producing system and metabolism differed in abundance between HCC and healthy control microbiomes. 35
Experimental animal model supporting the role of the gut microbiota in hepatocarcinogenesis.
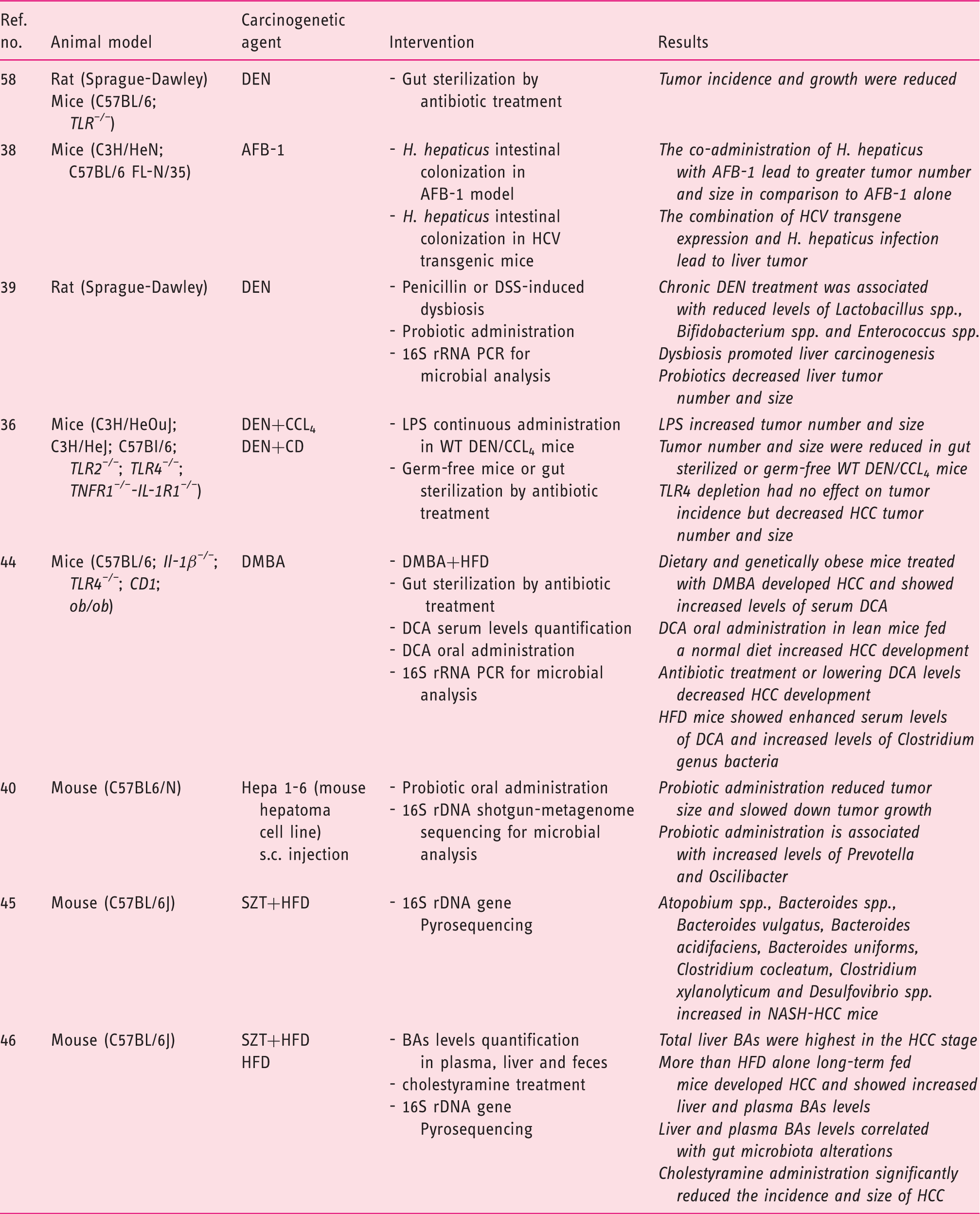
HCC: hepatocellular carcinoma; TLR: toll-like receptor; DEN: diethylnitrosamine; AFB: alfatoxin; DSS: dextran sulfate sodium; CCl4: carbon tetrachloride; CD: choline-deficient diet; LPS: lipopolysaccharide; WT: wild type; DMBA: dimethylbenz(a)anthracene; HFD: high-fat diet; DCA: deoxycholic acid; SZT: streptozotocin; BA: bile acid; LCA: lithocholic acid; TDCA: taurochenodeoxycholic acid PCR: polymerase chain reaction; rRNA: ribosomal RNA; rDNA: ribosomal DNA; s.c.: subcutaneous; TNFR1: tumor necrosis factor receptor 1; IL-1R1: interleukin 1 receptor type 1.
The presence of Helicobacter spp. in human samples of HCC suggested the direct involvement of bacteria in liver carcinogenesis. 37 The colonization of mouse intestine by Helicobacter hepaticus, in a model of aflatoxin-induced liver cancer, led to the development of tumors greater in number and size. 38 However, in mice harboring a full-length hepatitis C virus (HCV) transgene, H. hepaticus colonization resulted in greater tumor burden and incidence, but neither H. hepaticus nor HCV transgene expression alone were sufficient for liver cancer initiation. Notably, the increased risk of HCC was independent of H. hepaticus translocation to the liver, indicating that the bacteria may induce HCC from its niche into the intestinal mucosa layer. 38 The analysis of the fecal and cecal microbiota in a rat model of chemically induced hepatocarcinogenesis revealed a profound dysbiosis with a decreased abundance of Lactobacillus spp., Bifidobacterium spp. and Enterococcus spp. and a higher level of gram-negative bacteria such as E. coli, Atopobium, Collinsella, Eggerthella and Coriobacterium with a concomitant increase in serum LPS levels. 39 The induction of intestinal dysbiosis by penicillin or dextran sulfate sodium (DSS) significantly promoted liver carcinogenesis, while the restoration of dysbiosis by probiotics decreased LPS serum levels and liver tumor number and size. 39
The beneficial effects of probiotics on liver immune differentiation and carcinogenesis were confirmed in a mouse model of HCC by using a novel probiotic mixture (Prohep). The administration of Prohep slowed down tumor growth and decreased tumor volume by 40% with respect to untreated mice. An increased level of Prevotella and Oscilibacter in the fecal microbiota of probiotic-treated mice suggested that the anti-inflammatory effect of these beneficial bacteria could positively affect liver carcinogenesis. It is likely that the promotion of a T regulatory (T-reg) cell immune-response by bacteria-derived metabolites decreases the migration of T helper 17 (Th17) cells to the liver. 40 Indeed, previous studies found an increased level of Th17 cells in the tumor and peri-tumor tissue suggesting their implication in liver carcinogenesis.41–43 Overall these data draw a potential new pathway of liver carcinogenesis based on a “gut microbiota-liver axis” paving the way to new possible strategies for the prevention and treatment of liver cancer.
The obesity-associated gut microbiota is characterized by increased levels of secondary BA deoxycholic acid (DCA), known to promote hepatocarcinogenesis. 44 Genetically or high-fat diet (HFD)-induced obesity was associated with higher levels of DCA and a higher incidence of HCC in mice treated with the chemical carcinogen dimethylbenz(a)anthracene (DMBA). Opposite, lean mice fed a normal diet and treated with DMBA did not develop liver cancer. Furthermore, vancomycin administration or lowering DCA levels reduced HCC development. 44 The analysis of the fecal microbiota of HFD-fed mice revealed an increase in the relative abundance of microbes belonging to the Clostridium genus that includes such bacteria producing DCA. 44 Recent data also reported that the intrahepatic retention of hydrophobic BAs including DCA were significantly increased in a streptozotocin- and HFD-induced NASH/HCC mouse model. 45 More than half of the HFD-fed mice developed HCC with increased liver and plasma levels of Bas, while lowering BAs by cholestyramine feeding significantly decreased the incidence of liver cancer. The same authors, in the same animal model of NASH/HCC, reported a marked increase in the fecal content of Atopobium spp., Bacteroides spp., Bacteroides vulgatus, Bacteroides acidifaciens, Bacteroides uniformis, Clostridium cocleatum, Clostridium xylanolyticum and Desulfovibrio spp., paralleling the LPS serum levels and the progression of liver disease. 46
Animal model studies provided the biological plausibility for the involvement of gut microbes in hepatocarcinogenesis and the rationale for hypotheses to be addressed by clinical research. Nevertheless, the extrapolation of data from animals to humans can be hampered by how well the animal’s phenotype mimics that of the human disease and its progression. This is particularly the case of liver cancer, which recognizes different etiologic factors, each one likely linked to specific molecular pathways of cancer initiation and progression. On the other side, given the scarcity and infancy of data from humans, well-designed and powered studies are advisable to gain insight on this fascinating topic.
Concluding remarks
Although convincing data established a deep alteration of gut microbiota in liver cirrhosis and its complications, with an increase of non-autochthonous taxa and a decrease of autochthonous taxa, we are still far from delineating a microbiota signature of the diseases. Experimental animal models suggest a promoting effect of the gut microbiota-driven inflammation in hepatocarcinogenesis but human studies are still lacking. Well-powered and designed studies based on standardized methodologies are advisable although the long-lasting natural history of the liver diseases is one of the major drawbacks in planning prospective clinical trials that would fill the gap in the knowledge on the pathogenetic role of gut microbiota in liver diseases. Furthermore, future studies should overcome the mere exploration of the microbiome composition and include metabolomic analysis taking into account that, beyond bacteria, viruses and mycetes are active components of the gastrointestinal microbial ecosystem.
The rapidly evolving landscape of microbiological technologies will likely furnish in the future the real picture of the complexity of the gut microbiome and the mechanisms underlying the development and progression of liver diseases. Achieving these goals would allow the consideration of new pathogenetic pathways to prevent and treat these devastating diseases.
Footnotes
Declaration of conflicting interests
None declared.
Ethics approval
Not applicable.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Informed Consent
Not applicable.