
Abstract
The frequency of fibrosing Crohn’s disease (CD) is significant, with approximately 40% of CD patients with ileal disease developing clinically apparent strictures throughout their lifetime. Although strictures may be subdivided into fibrotic, inflammatory, or mixed forms, despite immunosuppressive therapy in CD patients in the form of steroids or immunomodulators, the frequency of fibrostenosing complications has still remained significant. A vast number of genetic and epigenetic variables are thought to contribute to fibrostenosing disease, including those that affect cytokine biology, and therefore highlight the complexity of disease, but also shed light on targetable pathways. Exclusively targeting fibrosis may be difficult, however, because of the relatively slow evolution of fibrosis in CD, and the potential adverse effects of inhibiting pathways involved in tissue repair and mucosal healing. Acknowledging these caveats, cytokine-targeted therapy has become the mainstay of treatment for many inflammatory conditions and is being evaluated for fibrotic disorders. The question of whether anti-cytokine therapy will prove useful for intestinal fibrosis is, therefore, acutely relevant. This review will highlight some of the current therapeutics targeting cytokines involved in fibrosis.
“Regulatory” cytokines
Transforming growth factor beta (TGFβ)
Cytokine and drug targets in fibrosis.
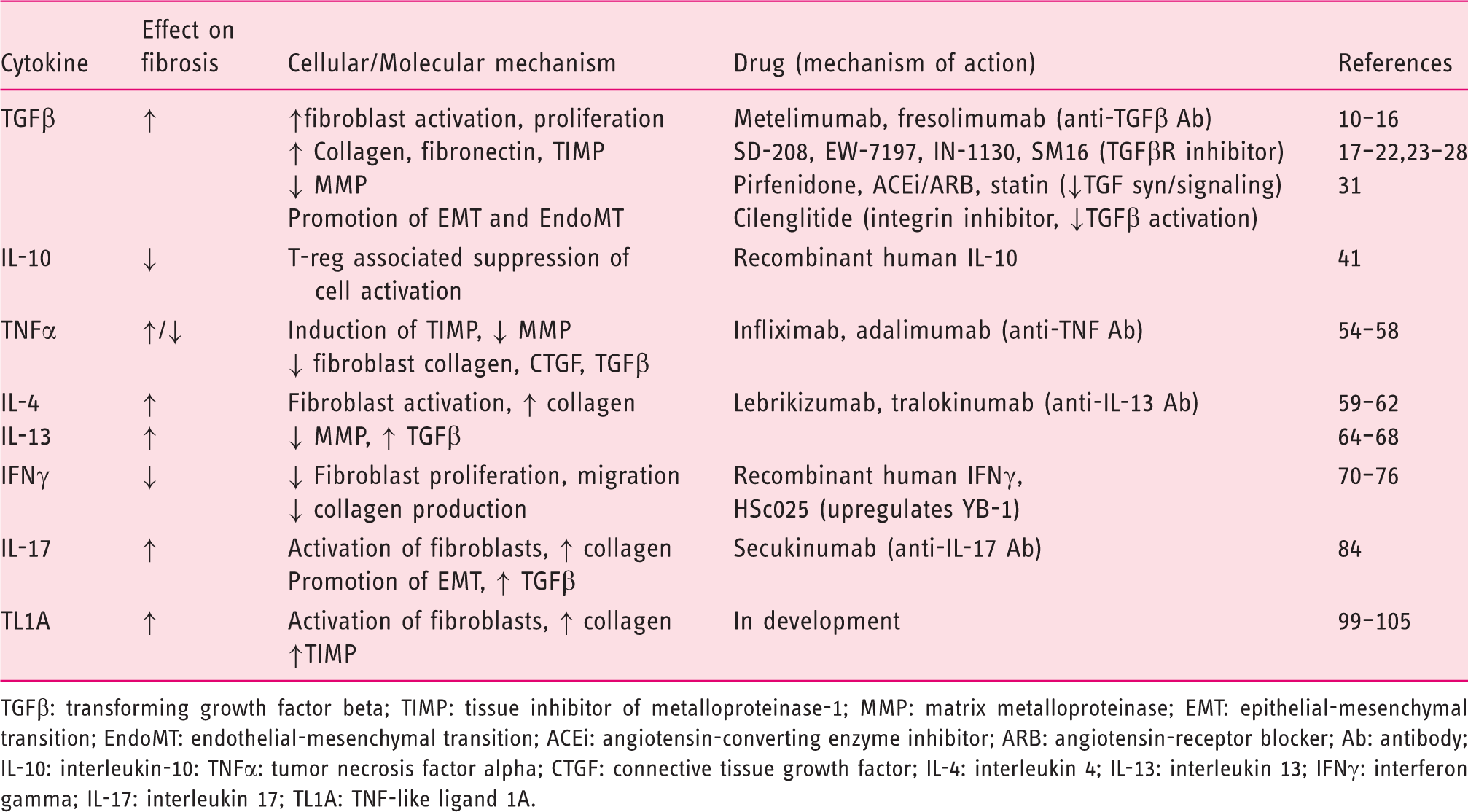
TGFβ: transforming growth factor beta; TIMP: tissue inhibitor of metalloproteinase-1; MMP: matrix metalloproteinase; EMT: epithelial-mesenchymal transition; EndoMT: endothelial-mesenchymal transition; ACEi: angiotensin-converting enzyme inhibitor; ARB: angiotensin-receptor blocker; Ab: antibody; IL-10: interleukin-10: TNFα: tumor necrosis factor alpha; CTGF: connective tissue growth factor; IL-4: interleukin 4; IL-13: interleukin 13; IFNγ: interferon gamma; IL-17: interleukin 17; TL1A: TNF-like ligand 1A.
The three main isoforms of TGFβ: TGFβ1, TGFβ2, and TGFβ3, are secreted as latent precursor molecules containing a latency-associated peptide region (LAP), and complexed with latent TGFβ binding proteins (LTBP). The cytokine is active when LTBP is removed extracellularly via proteolytic cleavage by proteases such as plasmin or thrombin, or by interactions of LAP with other proteins such as thrombospondin-1 or integrins.
4
TGFβ signaling ensues through two receptors, TGFβR1 and TGFβR2, which form transmembrane serine/threonine kinase, hetero- or homo-dimeric complexes that induce phosphorylation of Smad 2 and Smad 3 proteins. Once phosphorylated, Smad 2 and 3 complexes with Smad 4, translocate to the nucleus, and activate transcription. Regulation of Smad 2/3 occurs via Smad 7, which prevents binding of Smad2/3 to the receptor complex. Signaling of TGFb is summarized in Figure 1. TGFβ can also signal through other pathways, however, including extracellular signal-regulated protein kinases 1 and 2 (ERK1/2), c-Jun N terminal kinase, p38 kinases and members of the Janus kinase/signal transducers and activators of transcription (JAK/STAT) family.4,9
Signaling of TL1A, TNFα, and TGFβ.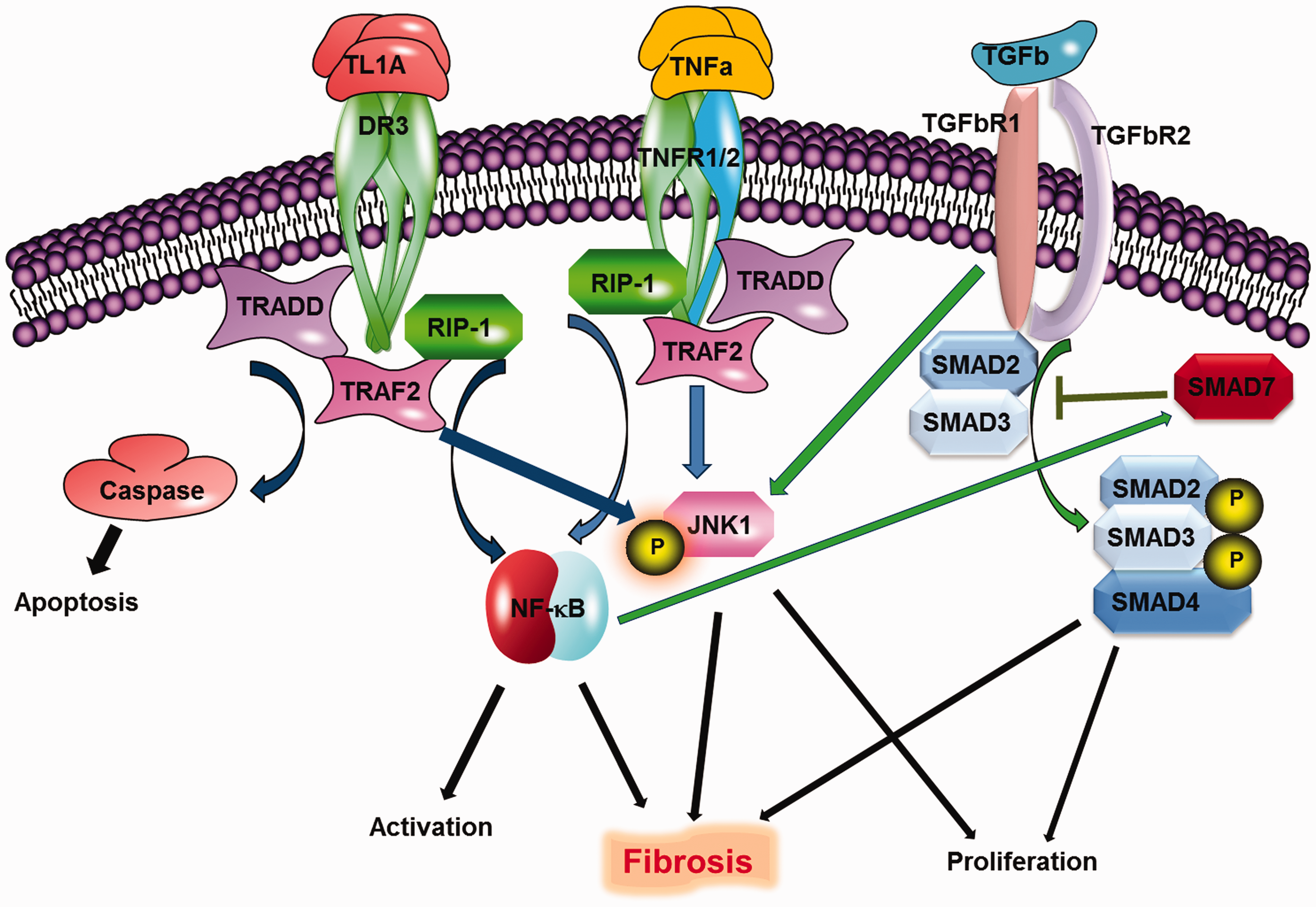
As mentioned above, in addition to its profibrotic effects, TGFβ is a potent immune modulator central to immune tolerance and development of innate and adaptive immunoregulatory cells. Thus, the global blockade of TGFβ might upset critical balances in immune homeostasis resulting in untoward effects, or perhaps be ineffective owing to simultaneous blockade of fibrogenic and regulatory functions. Several global TGFβ-blocking agents were either found to be ineffective or led to possible drug-associated mortality.10,11 While global blockade of TGFβ might be problematic, other strategies have focused on specific pathways in TGFβ signaling, synthesis, activation, or other downstream mediators and effects, with the hope of targeting TGFβ-driven fibrosis while sparing its immunomodulatory effects. Thus in light of this broad complexity of function, antagonizing an individual receptor, rather than the ligand itself, might be more attractive if it proves more efficacious and specific. Accordingly, blockade of TGFβR1 signaling by an injectable inhibitor (SD-208) was evaluated in two experimental animal models of intestinal fibrosis: anaerobic bacteria- and trinitrobenzensulphonic acid-induced colitis (TNBS). SD-208 reduced fibroblast activation, phosphorylation of Smad2 and Smad3 proteins, and intestinal wall collagen deposition in both models. 12 Similarly, more recent studies on blockade of TGFβR1 with oral inhibitors have demonstrated efficacy in animal models of renal fibrosis, carbon tetrachloride- or bile duct ligation-induced cirrhosis,13,14 pressure-overload-induced cardiac fibrosis, 15 and bleomycin-induced pulmonary fibrosis. 16 These agents are currently being investigated in oncologic trials, with pre-clinical testing ongoing for fibrotic disorders.
With regards to TGFβ synthesis, 5-methyl-1-phenyl-2-[1H]-pyridone (pirfenidone) is a small, orally active molecule that has demonstrated anti-fibrotic effects, in part via inhibiting synthesis of TGFβ. This agent has been efficacious in patients with, and experimental models of, pulmonary and renal fibrosis.17,18 Pirfenidone has been evaluated in randomized, double-blind, placebo-controlled clinical trials. Pirfenidone reduced the rate of decline in lung function as measured by changes in forced vital capacity or total lung capacity, as well as improved mortality.19,20 It has been approved in Europe and by the Food and Drug Administration (FDA) for treatment of idiopathic pulmonary fibrosis (IPF). Pirfenidone, however, has not been uniformly beneficial in all clinical trials; it had no clinical or histologic benefits in patients with myelofibrosis, 21 or primary sclerosing cholangitis, while being associated with increased adverse events. 22
Downregulating or decreasing production of TGFβ without adverse immunological effects has been demonstrated by two classes of medications currently in widespread use in primary care: HMG-CoA reductase inhibitors (statins) and antagonists of the renin-angiotensin system (RAS). As the primary mediator of the RAS, angiotensin may contribute to fibrogenesis via induction of TGFβ expression and promotion of collagen production. 23 With regards to intestinal fibrosis, early studies have reported that angiotensin is increased in the mucosa of Crohn’s disease (CD) patients. 24 In TNBS-induced colitis, administration of the angiotensin-converting enzyme (ACE) inhibitor (ACEi), captopril, or the angiotensin receptor blocker (ARB), losartan, reduced colonic inflammation and fibrosis via reduction in TGFβ1.25,26 Like antagonists of the RAS, statins may be of benefit with regards to fibrosis, in part, through decreasing expression of TGFβ. Simvastatin reduces TGFβ1 expression in human fibroblasts by inhibition of Smad 3 phosphorylation. 27 In TNBS-induced colitis, it had anti-fibrotic effects characterized by a dose-dependent decrease in the level of connective tissue growth factor (CTGF) and induction of apoptosis in fibroblasts. 28 Given the safety and ubiquity of RAS antagonists and statins, future prospective investigations will be feasible and determine if they are capable of favorably affecting fibrogenesis.
The activation of TGFβ from its latent precursor state serves as an important regulatory step in TGFβ signaling, which might be exploited as a therapeutic target. Strategies of integrin inhibition, most recently with vedolizumab, have proven effective with regards to inflammation in inflammatory bowel disease (IBD) and may affect fibrosis via their effects on TGFβ activation. As mentioned above, integrins, particularly those of the alphaV (αV)-type, can bind LAP and activate TGFβ. αVβ6 integrin is upregulated in various fibrotic disorders and its blockade has been effective in models of radiation- and bleomycin-induced pulmonary fibrosis, as well as liver fibrosis. 29 Similarly, αVβ3 integrin contributes to excess smooth muscle cell proliferation and hyperplasia in intestinal strictures of CD, 30 and cilengitide, an αVβ3 inhibitor, reduces the development of fibrosis in chronic TNBS-induced colitis. 31 Future studies will demonstrate if these, or the currently used integrin inhibitors, will have favorable effects with regards to fibrosis in IBD.
Another seemingly attractive option is the targeting of specific signaling molecules in the TGFβ cascade. This option might appear favorable, as it may focus on individual mediators of TGFβ signaling rather than broader targets such as TGFβ itself. Two such potential strategies are Smad 3 antagonism and Smad 7 agonism. Increased Smad 3 and decreased Smad 7 expression have been observed in intestinal strictures in CD. 32 Furthermore, in multiple animal models, loss of Smad 3 or increase in Smad 7 confers resistance to fibrosis in several organs.33–35 There has been recent focus on inhibition of Smad 7 in IBD via antisense oligonucleotides (and subsequent increase in Smad 3 transduction with potential TGFβ-mediated shift toward immune-regulation). This strategy may be troubling with regards to fibrogenesis. An ideal solution might be to clearly identify those patients that would be more prone to develop fibrotic/stricturing disease vs predominantly inflammatory pathology through functional, genetic, and epigenetic studies.
Interleukin-10 (IL-10)
IL-10 has a well-known role with regards to immune regulation as a prominent product of regulatory T cells and their effects on intestinal inflammation. 36 In contrast to TGFβ, however, IL-10 has been shown to inhibit fibrosis. Mice treated with IL-10 develop less liver and lung fibrosis when administered carbon tetrachloride or bleomycin.37,38 Similarly, IL-10 deficiency aggravates kidney inflammation and fibrosis in the unilateral ureteral obstruction mouse model. 39 With regards to human IBD, however, although polymorphisms in the IL-10 locus have been associated with IBD, 40 treatment of CD patients with recombinant IL-10 has not been significantly effective (Table 1). 41
“Inflammatory” cytokines
Tumor necrosis factor alpha (TNFα)
Like TGFβ, TNFα is a pleiotropic cytokine, classically considered proinflammatory with important immunomodulatory properties. A variety of cell types can elaborate TNFα, including activated macrophages, B cells, T cells, keratinocytes, and fibroblasts. Depending on the conditions, TNFα can trigger either pro-inflammatory or anti-inflammatory pathways by engaging one or both of two distinct transmembrane receptors: TNFR1, and TNFR2. In addition to its pro-inflammatory effects, TNFα may potentiate fibrosis via induction of TIMP-1 expression and reduction in MMP-2 activity and collagen degradation. 42 Treatments targeting TNFα are perhaps some of the most widely used anti-cytokine therapies for inflammatory disorders, but evidence for the role of these agents in preventing fibrosis is somewhat mixed. In some animal models of liver and renal fibrosis, TNF blockade reduced organ inflammation and fibrogenesis,43,44 but a recent clinical study investigating adalimumab for fibrotic kidney disease (FSGS) failed to meet its primary outcome. 45 An open-label pilot study in 16 systemic sclerosis patients demonstrated improvement in skin scores with reduction in collagen secretion noted from cultured lesional fibroblasts (Table 1).46–48
In contrast, there is evidence to suggest that TNFα is a potentially anti-fibrogenic cytokine and its blockade might consequently promote fibrosis. In some studies, TNFα can exhibit anti-fibrotic properties by reducing the expression of collagen and CTGF in dermal fibroblasts, 49 and via suppression of TGFβ signaling through nuclear factor (NF)-kappa (K) B induction of Smad 7 in other cell types. 50 Disparate effects may be cell specific and segregate at the level of the individual TNF receptors, as globally impaired signaling through TNFR1 accelerates wound-healing, increases collagen deposition, and angiogenesis at wound sites in TNFR1-deficient mice, 51 whereas impaired signaling in TNFR2-deficient intestinal myofibroblasts results in reduced cell proliferation and decreased collagen synthesis. 42
Consequently, with regards to intestinal fibrosis, the evidence for specific use of TNF antagonists as anti-fibrotic agents (as opposed to anti-inflammatory agents) has remained vague. In early reports of TNF blockade, obstructive complications were observed in some patients, with initial concerns that these agents may promote excessive fibrotic changes accompanying mucosal healing. In vitro studies, however, showed that TNF blockade decreased myofibroblast collagen production 52 in CD patients treated with infliximab. Later multivariable analyses from the observational Crohn’s Therapy, Resource, Evaluation, and Assessment Tool (TREAT) registry and the A Crohn’s Disease Clinical Trial Evaluating Infliximab in a New Long-term Treatment Regimen (ACCENT) I multicenter trial determined that, rather than TNF-antagonist use, disease duration, severity, location, and new corticosteroid use are factors associated with stricture formation. 53 Positive results have now been seen in a few patients with inflammatory or mixed stenosis,54,55 as well as small case series reporting intralesional injection of infliximab. 56 Data from population-based cohorts seem to suggest that these agents may reduce the need for surgery in the short term 57 with the rate of surgery ranging between 27% and 61% within the first five years after diagnosis before the introduction of biologics, and between 25% and 33% after the introduction of anti-TNF agents. 58 Indeed, anti-TNF agents are recommended to reduce the risk of postoperative recurrence after surgery. Discerning between unique anti-fibrotic effects in these cases and modification of the fibrotic program due to reduction in inflammation may be difficult.
T helper (Th)2 cytokines
The Th2 cytokines, interleukin-4 (IL-4) and interleukin (IL-13), have been implicated in fibrogenesis (Table 1). Both are elevated in fibrotic disease and promote fibroblast activation, proliferation, and collagen synthesis.59,60 For example, IL-4 is found at increased concentrations in the bronchoalviolar lavage of patients with IPF. 61 IL-4 also increases the expression of collagen in cultured hepatic fibroblasts. 62 Similarly, IL-13 is involved in many Th2-mediated diseases and has a role in fibrosis as well. Deriving from a common receptor subunit (IL-4Ralpha), IL-13 shares overlapping functions with IL-4. IL-13 signals by interacting with a complex receptor system composed of IL-4Ralpha and two IL-13 binding proteins, IL-13Rα1 and IL-13Rα2. IL-13 receptors are expressed on a vast array of cells, including human hematopoietic cells, endothelial cells, fibroblasts, multiple epithelial cell types, and smooth muscle cells. 63 Increased IL-13 messenger RNA (mRNA) was found in intestinal samples of fibrotic CD patients. Fibroblasts from these samples expressed elevated levels of IL13Rα1 and subsequently downregulated MMP in response to IL-13. 64 Importantly, however, elevated IL-13 production was not detected in ulcerative colitis (UC) or strictured CD, 65 questioning if anti-IL-13 therapy would be an appropriate strategy in IBD. In vivo inhibition of IL-13Rα2 expression reduced production of TGFβ1 in oxazolone-induced colitis and led to a marked decrease of collagen deposition in bleomycin-induced lung fibrosis. 66 Similarly, IL-13 blockade reduces experimental hepatic fibrosis. 67 In TNBS-induced colitis, inhibition of IL-13 signaling by administration of small interfering RNA targeting the IL-13Rα2, reduces fibrosis and expression of TGFβ. 68 Given the suggested experimental benefits of IL-13 antagonism, IL-13 antibodies such as lebrikizumab and tralokinumab are currently being evaluated for anti-fibrotic efficacy in pulmonary fibrosis (ClinicalTrials.gov identifiers: NCT01872689, NCT01629667). Given the benefit in pre-clinical investigations, clinical studies targeting the IL-13 or IL-13 receptor may be envisioned for fibrosis in CD.
Th1 cytokines
In contrast to pro-fibrotic cytokines produced by Th2 cells, Th1 cells, through production of interferon gamma (IFNγ), have opposing anti-fibrotic effects. IFNγ has been shown to inhibit fibroblast proliferation and migration. 69 IFNγ signaling was shown to suppress the production of TGF-b via Y box-binding protein (YB-1), and an orally administered compound that promotes nuclear translocation of YB-1 resulted in the improvement of murine liver fibrosis and TNBS-induced murine chronic colitis.70–72 Several other models have demonstrated the potent anti-fibrotic activity of IFNγ. In the case of schistosomiasis-induced fibrosis, treatment with IFNγ reduces collagen deposition associated with chronic granuloma formation. 73 Similar results were obtained in models of pulmonary and kidney fibrosis.74,75 These outcomes were not replicated in human studies, however. A randomized trial of subcutaneously injected recombinant IFNγ did not demonstrate improvement in survival of patients with IPF (Table 1). 76
Th17 cytokines
The family of interleukin-17 (IL-17) cytokines is composed of IL-17A-F, which act through the IL-17 receptor. Early evidence suggested that a main function of IL-17 is the promotion of chemokine production for granulocyte activation and increasing inflammation. 77 With regards to pro-fibrotic effector functions, IL-17 stimulates activation pathways in human colonic myofibroblasts 78 and maintains fibrotic activity in various other cell types including stellate cells 79 and lung epithelial cells. 80 Treatment of mice with anti-IL-17A monoclonal antibody administered after the onset of myocarditis abrogates cardiac fibrosis and preserves ventricular function. 81 Similarly, IL-17A increases the synthesis and secretion of collagen and promotes the epithelial-mesenchymal transition in alveolar epithelial cells in a TGFβ1-dependent manner. Neutralization of IL-17A promotes the resolution of bleomycin-induced acute inflammation, attenuates pulmonary fibrosis, and increases survival in this model. 80 In IBD, however, IL-17’s contribution to disease pathogenesis is complex as both human and animal data suggest a dual inflammatory and protective role. With regards to its role in intestinal fibrosis, in vitro intestinal samples from fibrosing CD patients express elevated levels of IL-17A and IL17-stimulated myofibroblasts from CD strictures generate more collagen and TIMP-1. 82 The role of IL-17 concerning clinical disease development in animal models of IBD has yielded disparate results depending on the model used. 83 In a clinical trial of patients with inflammatory CD, blockade of IL-17A by administration of the anti-IL-17A antibody, secukinumab, failed to meet its primary endpoint (Table 1). 84 Post hoc analysis identified that a subgroup of patients who responded to IL-17 blockade carried a TNFSF15 (rs4263839) single nucleotide polymorphism (SNP). The potential functional consequences of this allele include elevated production of TNF-like ligand 1A (TL1A) protein. Under TL1A-upregulated conditions in adoptive transfer-induced colitis, IL-17A deficiency ameliorated colonic inflammation via reducing Th1 and Th9 effector responses while enhancing regulatory responses. 85 Thus, there exists a subset of patients (those that overexpress TL1A owing to, for example, a TNFSF15 variant) who could potentially benefit from IL-17 blockade. Given the potential profibrotic role of TL1A overexpression in this subset of patients who have a propensity toward fibrostenosis (as discussed below), IL-17 blockade may have a positive impact on fibrosis, as well as inflammation.
TL1A
TL1A (a protein encoded by TNFSF15) is a member of the TNF superfamily that binds to death domain receptor 3 (DR3, also known as TNFRSF25), expressed on a variety of cell types.86–88 Modulating an array of immune responses, TL1A can be expressed by endothelial cells induced by interleukin-1β (IL-1β) and TNFα, macrophages and dendritic cells in response to Toll-like receptor stimulation, as well as in some lymphoid lineage cells.89–92 The role of TL1A in fibrosis is further described below and summarized in Table 1.
Much of the previous literature has focused on the TL1A-DR3 pathway with regards to immune function with recent evidence now suggesting its importance concerning fibrosis. As DR3 shares homology with TNFR1, consequently, like with other TNF receptors, developmental, immunoregulatory, and pro-inflammatory effects have been described. DR3 activation of NF-KB in human cell lines upregulates cellular inhibitor of apoptosis 2 (c-IAP2), an NF-KB-dependent anti-apoptotic protein, which protects against apoptosis. 93 Conversely, however, DR3 in embryonic cells can induce Fas-associated death domain protein (FADD)- and caspase-8-dependent apoptosis, and early work on DR3-deficient mice demonstrated that it is required for negative selection in the thymus.94,95 Despite its initially recognized role as a pro-apoptotic receptor, DR3 has been shown to be upregulated on Th17 cells, and TL1A-DR3 interaction promotes T cell expansion and cytokine production during immune responses.96–98 Along these lines, the pro-inflammatory effects of TL1A-DR3 binding likely contribute to this pathway’s effect on fibrosis. More direct evidence has demonstrated that DR3 is an important receptor for fibroblast development, maturation, and function. Indeed, DR3 is expressed on human and mouse primary intestinal fibroblasts, and DR3-deficient mice display reduced numbers of colonic fibroblasts. DR3 deletion, in addition to conferring developmental/proliferative deficiencies in intestinal fibroblasts, also results in reduced fibroblast activation (as evidenced by decreased expression of alpha smooth muscle actin) and expression of collagen induced by TL1A stimulation. 99
Specifically with regards to the TL1A-DR3 pathway in IBD, a TNFSF15 haplotype is associated with higher TL1A expression, increased risk of CD, intestinal fibrostenosis, and greater need for surgery.100–102 In mice, constitutive TL1A overexpression causes spontaneous ileitis with increased collagen deposition.103,104 Under colitogenic conditions induced by chronic dextran sulfate sodium (DSS) treatment or adoptive T-cell transfer, increased inflammation, fibrosis, and fibrostenotic lesions in the gut are seen. 105 These results support the role of TL1A in induction of intestinal inflammation and suggest its contribution to fibrogenesis in the gut. The potential for TL1A as a therapeutic target in intestinal fibrosis was demonstrated in a recent study evaluating the effect of anti-TL1A antibodies (Ab) in chronic DSS and adoptive T-cell transfer models of IBD. Treatment with neutralizing TL1A Ab attenuated disease and reversed colonic fibrosis. Additionally, TL1A blockade reduced the number of fibroblasts and myofibroblasts in colonic cell isolates and lowered expression of CTGF, TGFβ1 and insulin-like growth factor 1 (IGF-1). 99 Current studies are underway to evaluate the direct and indirect contributors in the TNFSF15/TL1A pathway to intestinal fibrosis.
Concluding remarks and future directions
Cytokine targeting has proven to be robustly effective in targeting inflammation in IBD. Given the pleiotropy of many cytokines, cytokine targeting with regards to intestinal fibrosis has been challenging. Moreover, the genetic variability present across patient populations may result in different pathogenesis of disease with regards to cytokine pathways, and thus broad cytokine targeting may result in disparate rates of response. Indeed, this has been observed with regards to anti-TNF agents in terms of inflammation and may be one source of failure of some clinical trials with newer anti-cytokine agents. A potential future approach to overcome this difficult aspect of cytokine-targeting may require careful selection of patients based on genetic or biochemical characteristics. Indeed, as noted above, post hoc analysis of the trial with anti-IL17 identified that a subgroup of patients who responded to IL-17 blockade carried a TNFSF15 (rs4263839) SNP. Subsequent work in animal models suggested that under elevated levels of TL1A conditions, IL-17 blockade could be beneficial. Thus, patient selection for such genetic or biochemical traits as candidates for a specific anti-cytokine treatment may result in greater success with these agents. Additionally, there are promising lines of targets used for other fibrotic conditions that may be of benefit in CD and warrant investigation. Given the variables that contribute to fibrostenosis in CD, targeting of multiple points in the fibrotic pathway, including the cytokines themselves, may be an option. Future investigations into novel fibrogenic molecules and pathways may lead to additional and more selective therapeutic targets as well as the identification of specific patient groups that could best benefit from such individualized treatment.
Footnotes
Acknowledgment
The authors thank Erica Flores for assistance in formatting and gathering references.
Funding
This work is supported by the National Institutes of Health (NIH) T32 DK07180-40 (NJ), Specialty Training and Advanced Research (STAR) Program at UCLA (NJ), NIH R01 DK056328-16 (NJ, SRT and DQS), NIH K08 Career Development Award DK093578 (DQS), and the F. Widjaja Foundation Inflammatory Bowel & Immunobiology Research Institute (NJ, SRT and DQS).
Conflict of interest
None declared.