
Abstract
Background
Sphingosine kinase 1 (SphK1)/sphingosine-1-phosphate (S1P)/sphingosine-1-phosphate receptors (S1PRs) signaling plays a key role in inflammatory responses. Lei et al. showed that SphK1 inhibition presented a hepatoprotective effect on acute liver damage via decreasing hepatic high-mobility group box 1 (HMGB1) cytoplasmic translocation.
Objective
We aim to determine whether SphK1 or S1PRs inhibition improves lipopolysaccharide (LPS)/D-galactosamine (GalN)-induced acute liver failure by inhibiting the mitogen-activated protein kinases (MAPKs) pathway.
Methods
A mouse model of acute liver failure was induced by LPS/GalN. Male C57BL/6J mice (6–8 weeks) were randomly distributed into five groups: control group, LPS/GalN group, SphK1 inhibition group (LPS/GalN+SKI-5c), S1PR1 inhibition group (LPS/GalN+W146), and S1PR3 inhibition group (LPS/GalN+CAY10444).
Results
We confirmed the findings of Lei et al. that hepatic SphK1 expression was upregulated; serum transaminase activity (AST, ALT), as well as serum TNF-α and IL-6, were decreased by SphK1 inhibition. We further showed that the expression of S1PR1 and S1PR3 was augmented in response to LPS/GalN. SphK1 inhibition improves hepatic hemorrhage, and the activities of hepatic caspase-3 and myeloperoxidase (MPO). Furthermore, the activation of the MAPKs family (JNK, ERK and p38) was suppressed by SphK1 inhibition. However, S1PR1 or S1PR3 inhibition did not protect the mouse against liver damage, though S1PR1 or S1PR3 inhibition reduced serum TNF-α and IL-6, and partially attenuated the phosphorylation of the MAPKs signaling.
Conclusions
SphK1 inhibition improves LPS/GalN-induced liver injury by inhibiting activation of MAPKs signaling.
Keywords
Introduction
Acute liver failure is primarily characterized by encephalopathy, coagulopathy, hepatic metabolic dysfunction and high mortality. 1 The main causes of acute liver failure are virus infection and drug-associated liver injury. 1 In addition, sepsis may also cause acute hepatic failure. However, the signaling-associated pathogenesis of sepsis-induced liver failure is not completely understood.
Sphingosine kinase 1 (SphK1), one of the two SphK isoenzymes, results in the catalytic synthesis of sphingosine-1-phosphate (S1P). S1P not only acts as a signaling molecule in the cytoplasm but also may motivate S1P receptors (S1PRs) to participate in a signaling pathway outside of the cell. Elevated levels of SphK1 messenger RNA (mRNA) and protein were detected in lipopolysaccharide (LPS)-stimulated macrophages 2 and microglia. 3 Furthermore, the transcription of TNF-α, IL-1β and inducible nitric oxide synthase (iNOS) was attenuated as a result of the inhibition of SphK1. 3 Lufrano et al. demonstrated that SphK1 participates in LPS-induced inflammation in aged rats. 4 Specific S1PRs play a role in the mechanism of inflammation as well.5,6 Overall, SphK1/S1P/S1PRs signaling leads to the pathophysiology of inflammatory responses.
Lei et al. demonstrated that SphK1 inhibition improves acute liver damage by attenuating hepatic high-mobility group box 1 (HMGB1) cytoplasmic translocation. 7 However, whether inhibition of SphK1 and its receptors (S1PRs) participates in the pathogenesis of sepsis-induced liver failure remains unclear.
In our study, LPS/GalN was administered intraperitoneally to induce a mouse model of acute liver failure according to a previous report. 8 We examined whether SphK1/S1P/S1PRs signaling was stimulated in LPS/GalN-induced hepatitis. We also explored the effect of SphK1 or S1PRs inhibition on acute liver failure and the underlying mechanism.
Methods
Animals
Male C57BL/6J mice (6–8 weeks) were housed under general conditions (23 ± 2℃, 12-hour light–dark cycle) with standard chow and water. All of the procedures involving the animals were in accordance with the policies of the Animal Care and Use Committee of the Medical School of Jinling Hospital.
Experimental protocol
A mouse model of clinical sepsis-associated liver failure has usually been induced by LPS/GalN. 9 Therefore, we administered to the mice intraperitoneal 50 µg/kg LPS (Escherichia coli 0111:B4, Sigma-Aldrich) and 1 g/kg D-gal (Sigma-Aldrich). 8 To evaluate the influence of SphK1 inhibition on acute liver failure, 2 mg/kg of the selective SphK1 inhibitor SKI-5c 10 (Sigma-Aldrich) was given intraperitoneally 11 30 minutes before the LPS/GalN injection. 12 To examine the effects of selective S1PRs inhibition on acute liver failure, the animals received an intraperitoneal injection of 0.1 mg/kg of the specific antagonist W146 (Cayman Chemical) or the specific S1PR3 antagonist CAY10444 (Cayman Chemical) 10 minutes before and 30 minutes after the LPS/GalN injection. 13
The animals were randomized into the following five groups: control group, LPS/GalN group, SphK1 inhibition group (LPS/GalN+SKI-5c), S1PR1 inhibition group (LPS/GalN +W146), and S1PR3 inhibition group (LPS/GalN +CAY10444).
Sample collection
Liver tissues were separated six hours after the LPS/GalN administration. Serum was collected two hours and six hours after the LPS/GalN injection. These samples were stored at −80℃ until detection.
Western blotting
In brief, the proteins were separated using radioimmunoprecipitation assay (RIPA) buffer (Beyotime, Nanjing, China) with proteinase inhibitor cocktail tablets (Roche, Germany). Then, the proteins were transferred to polyvinylidene difluoride (PVDF) membranes. After blocking with 5% skimmed milk powder diluting in 0.1% Tween-20, the membranes were incubated at 4℃ overnight with the appropriate primary antibody, i.e. SphK1 (Cell Signaling Technology), S1PR1 (Abcam), S1PR3 (Abcam), p-c-jun N-terminal kinase (p-JNK) (Cell Signaling Technology), p-extracellular signal-regulated kinase (p-ERK) (Cell Signaling Technology), p-p38 (Cell Signaling Technology) or glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Abcam).
Liver histopathology
The livers were fixed in 4% formalin and embedded in paraffin and cut into 5-µm sections. Then, hematoxylin-eosin (H&E) was used to stain the sections.
Measurement of transaminase
The serum enzymatic activities of alanine transaminase (ALT) and aspartate transaminase (AST) were determined using commercial kits (Nanjing Jiancheng Bioengineering Institute, Nanjing, China).
Serum levels of tumor necrosis factor-alpha and interleukin-6 assay
The serum levels of tumor necrosis factor-alpha (TNF-α) and interleukin-6 (IL-6) were detected using commercial kits (Biosky, Nanjing, China).
Analysis of caspase-3 and myeloperoxidase activity in the liver
The enzymatic activities of caspase-3 and myeloperoxidase (MPO) in the liver were measured by assay kits (Genmed, Shanghai, China).
Statistical analysis
A one-way analysis of variance (ANOVA) followed by Bonferroni post hoc test was used to compare the difference between the groups using SPSS 19.0 software. If the variances were unequal, a log transformation was performed before one-way ANOVA. If the variances were still unequal after a log transformation, a non-parametric test with Kruskal-Wallis post hoc test was performed. Data were presented as the mean ± SD. A p < 0.05 was considered statistically significant.
Results
Upregulated expression of SphK1 and S1PR1,3 in LPS/GalN-treated mice
Previous reports have showed that SphK1 and specific S1PRs are linked with various inflammatory responses.2,3,14 Therefore, we evaluated the expression of SphK1 and S1PRs in an acute liver failure mouse model. We found that SphK1 expression in the LPS/GalN group was much higher than in the control group (Figure 1(a), p < 0.01). Elevated hepatic S1PR1 and S1PR3 protein expression was also notably detected in the LPS/GalN animals compared with the control group (Figure 1(b) and (c), p < 0.01, respectively).
Protein levels of SphK1, S1PR1 and S1PR3 are upregulated by LPS/GalN. Western blotting was used to determine the expression of hepatic SphK1 (a), S1PR1 (b) and S1PR3 (c) after LPS/GalN administration. The expression of each protein was indeed decreased by its specific inhibitor. The difference between groups was analyzed using one-way ANOVA with Bonferroni post hoc test. Data were presented as the mean ± SD, n = 3; **p < 0.01 vs control group. SphK1: sphingosine kinase 1; S1PR1: sphingosine-1-phosphate receptor 1; S1PR3: sphingosine-1-phosphate receptor 3; LPS: lipopolysaccharide; GalN: D-galactosamine; ANOVA: analysis of variance.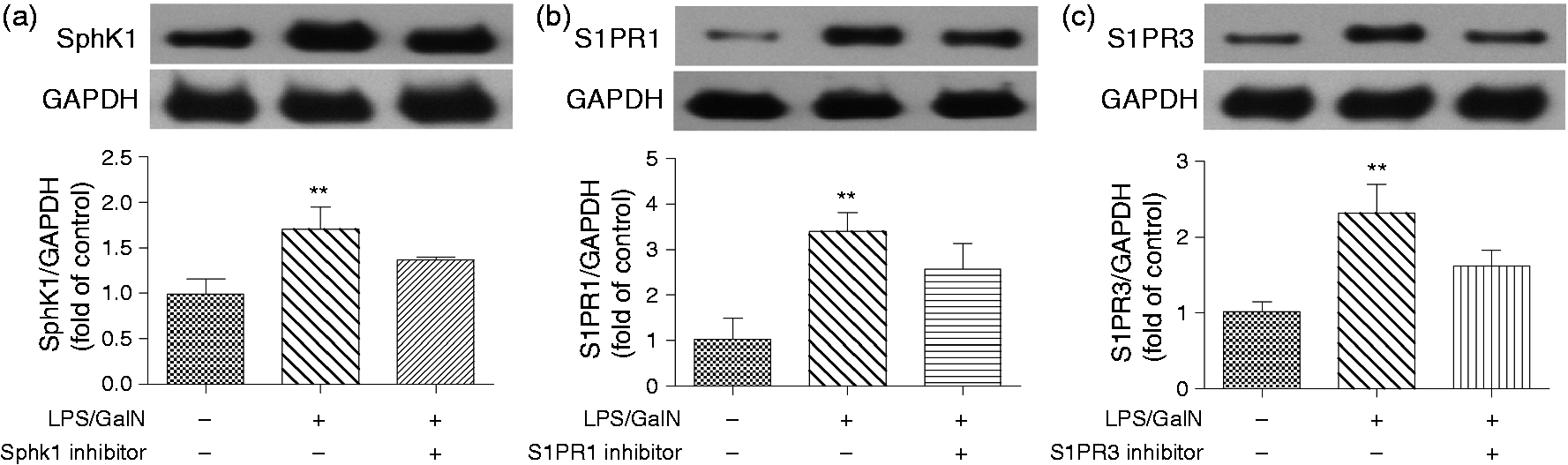
To further explore the role of SphK1-S1PRs in the LPS/GalN-induced liver failure, mice were administered specific antagonists for SphK1, S1PR1 or S1PR3 according to previous reports. Figure 1 shows that the expression of each protein was downregulated by the administration of its selective inhibitor.
Hepatic hemorrhage in histopathology was improved by SphK1 inhibition
Changes in hepatic histopathology were evaluated six hours after LPS/GalN administration. Histopathology indicated that hepatic hemorrhage in the LPS/GalN group was more severe than in the control group (Figure 2, p < 0.001). In contrast to the LPS/GalN group, the SphK1 antagonist significantly alleviated the severity of hepatic hemorrhage (Figure 2, p < 0.001). Liver histopathology was not markedly altered by S1PR1 or S1PR3 inhibitor treatment.
SphK1 inhibition improves hepatic hemorrhage in histopathology. Changes in hepatic histopathology were evaluated six hours after LPS/GalN administration with or without specific inhibitor for SphK1, S1PR1 or S1PR3. Quantification of hepatic hemorrhage is presented as a percentage of total area in chart. The difference between groups was analyzed using one-way ANOVA with Bonferroni post hoc test. Data were expressed as the mean ± SD, n = 6; ###p < 0.001 vs LPS/GalN group. SphK1: sphingosine kinase 1; S1PR1: sphingosine-1-phosphate receptor 1; S1PR3: sphingosine-1-phosphate receptor 3; LPS: lipopolysaccharide; GalN: D-galactosamine; ANOVA: analysis of variance.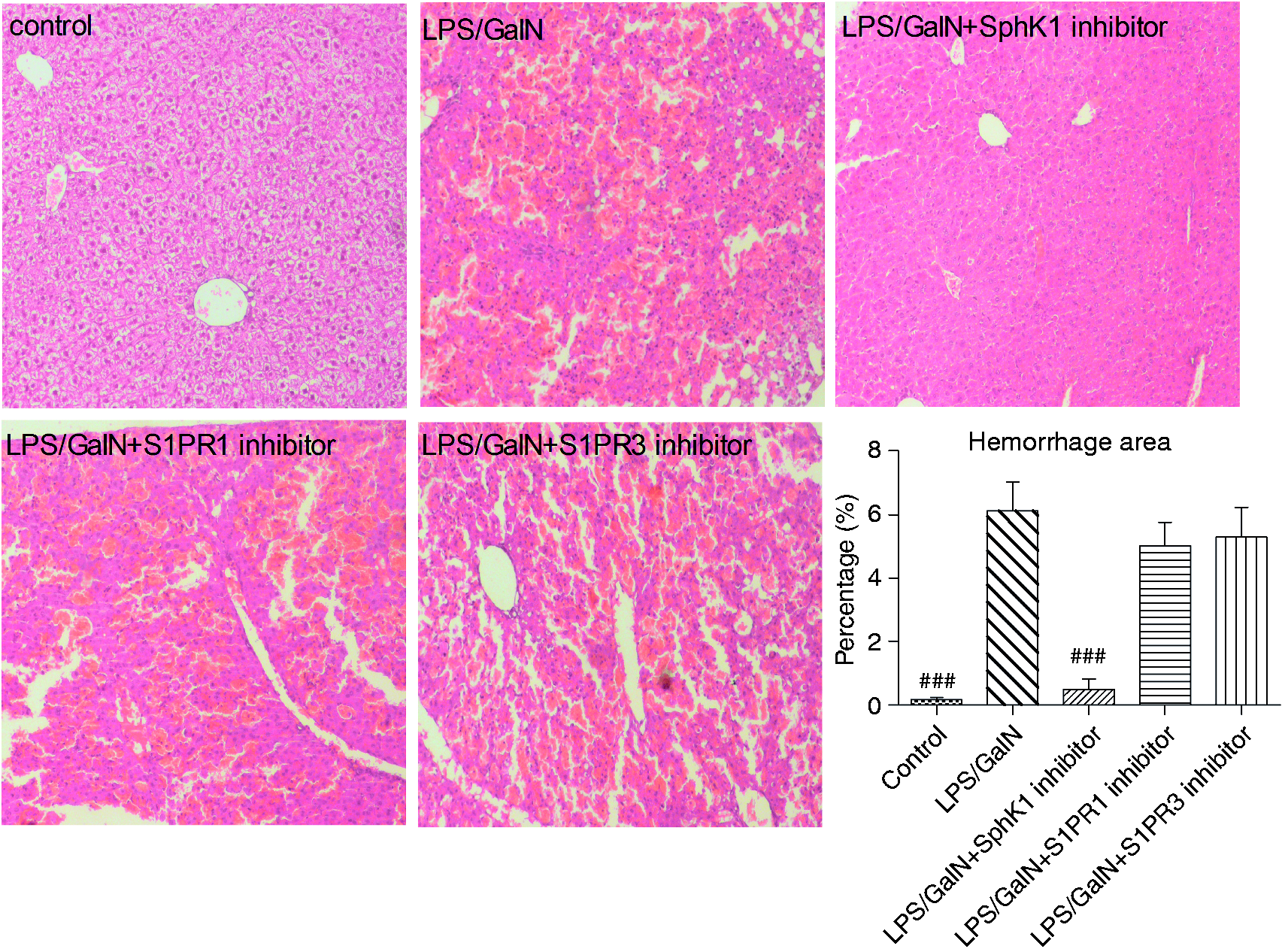
Serum transaminases activity was decreased by SphK1 inhibition
One of the main indexes of liver injury is serum transaminases. Serum AST and ALT activities were strongly increased by LPS/GalN, while with SphK1 inhibition, the activities were decreased (Figure 3). S1PR1 or S1PR3 suppression had no effect on these indexes (Figure 3).
SphK1 inhibition decreases the serum activities of AST and ALT. Serum transaminase activity (AST, ALT) was determined six hours after injection. AST values with activity analysis between groups were determined by non-parametric test with Kruskal-Wallis post hoc test. The variance of ALT values was not equal; a log transformation was performed before one-way ANOVA with Bonferroni post hoc test. Data were expressed as the mean ± SD, n = 10; ##p < 0.01, ###p < 0.001 vs LPS/GalN group. SphK1: sphingosine kinase 1; S1PR1: sphingosine-1-phosphate receptor 1; S1PR3: sphingosine-1-phosphate receptor 3; LPS: lipopolysaccharide; GalN: D-galactosamine; ANOVA: analysis of variance; AST: aspartate aminotransferase; ALT: alanine transaminase.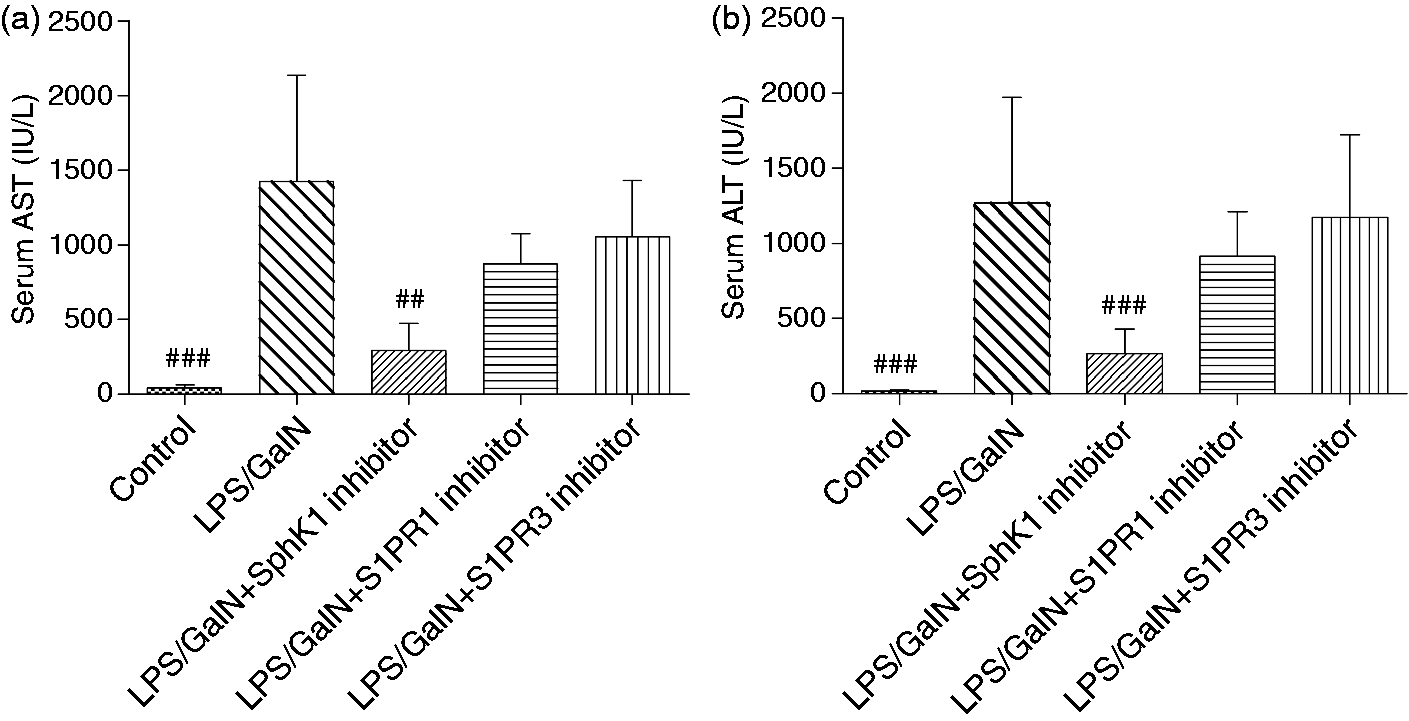
SphK1 inhibition reduced serum concentrations of TNF-α and IL-6
Inflammatory cytokines, especially TNF-α, have been known to be important in acute liver failure, and TNF-α and IL-6 are secreted at the early stage of the disease. Therefore, we measured the serum levels of TNF-α and IL-6 two hours after LPS/GalN administration. As indicated in Figure 4, serum TNF-α and IL-6 were strikingly increased in the LPS/GalN animals, whereas they were attenuated by the SphK1 inhibitor treatment. To our surprise, the inhibition of S1PR1 or S1PR3 also decreased serum TNF-α and IL-6.
SphK1 or S1PRs inhibition attenuates serum concentrations of TNF-α and IL-6. The serum TNF-α analysis between groups was determined using one-way ANOVA with Bonferroni post hoc test. The serum IL-6 analysis between groups was evaluated by non-parametric test with Kruskal-Wallis post hoc test. Data were presented as the mean ± SD, n = 10; ##p < 0.01, ###p < 0.001 vs LPS/GalN group. SphK1: sphingosine kinase 1; S1PR1: sphingosine-1-phosphate receptor 1; TNF-α: tumor necrosis factor alpha; IL-6: interleukin 6; LPS: lipopolysaccharide; GalN: D-galactosamine; ANOVA: analysis of variance.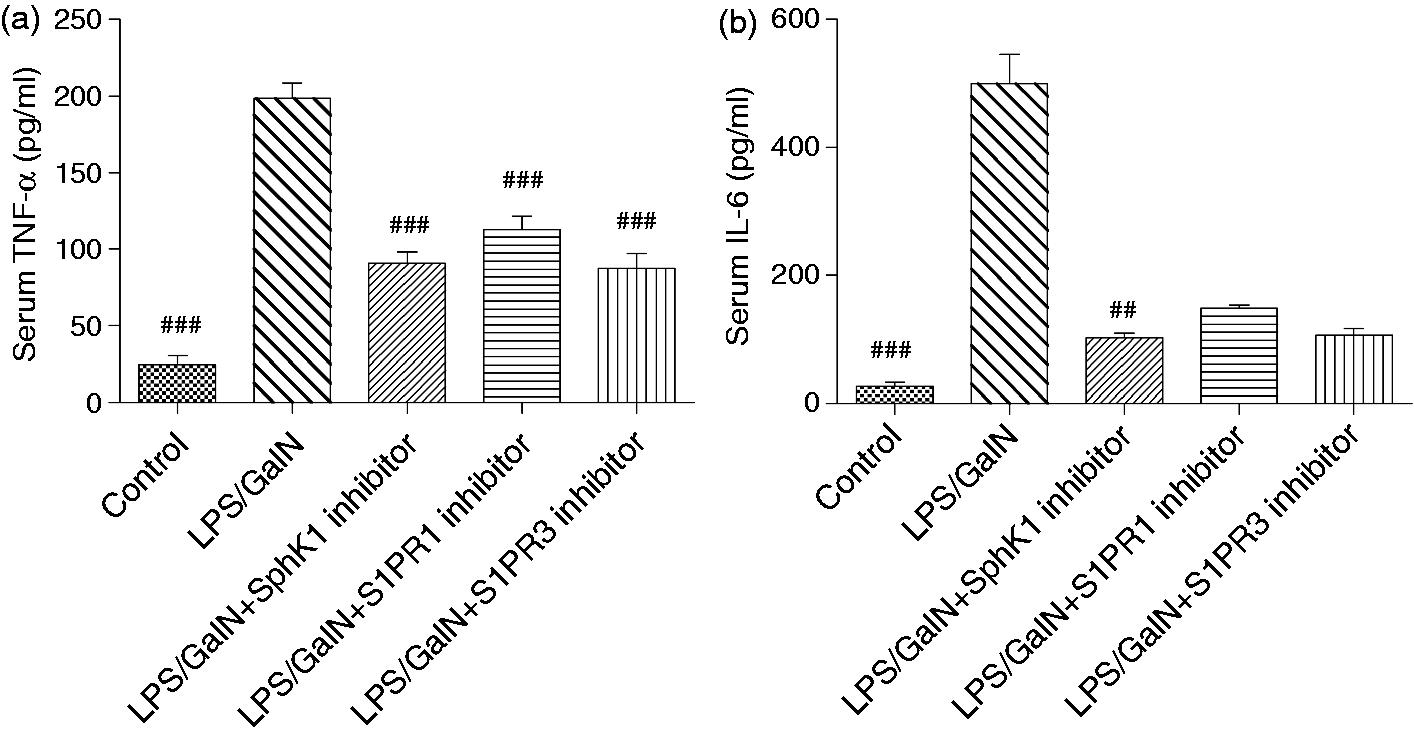
SphK1 inhibition attenuated hepatic caspase-3 and MPO activities
To further explore the effects of SphK1 and S1PRs inhibition on liver failure, we detected caspase-3 and MPO activities in the liver. The data indicated that liver caspase-3 and MPO activities were facilitated in the LPS/GalN group and were downregulated after administration of the SphK1 inhibitor (Figure 5). The suppression of S1PR1 or S1PR3 had no significant effect on these indexes.
The activities of hepatic caspase-3 and MPO are reduced by SphK1 inhibition. The activities of caspase-3 and MPO in the liver were determined six hours after LPS/GalN administration. The caspase-3 activity analysis between groups was evaluated by non-parametric test with Kruskal-Wallis post hoc test. The variance of MPO values was not equal; a log transformation was performed before one-way ANOVA with Bonferroni post hoc test. Data were presented as the mean ± SD, n = 10; ##p < 0.01 vs LPS/GalN group. MPO: myeloperoxidase; SphK1: sphingosine kinase 1; LPS: lipopolysaccharide; GalN: D-galactosamine; ANOVA: analysis of variance.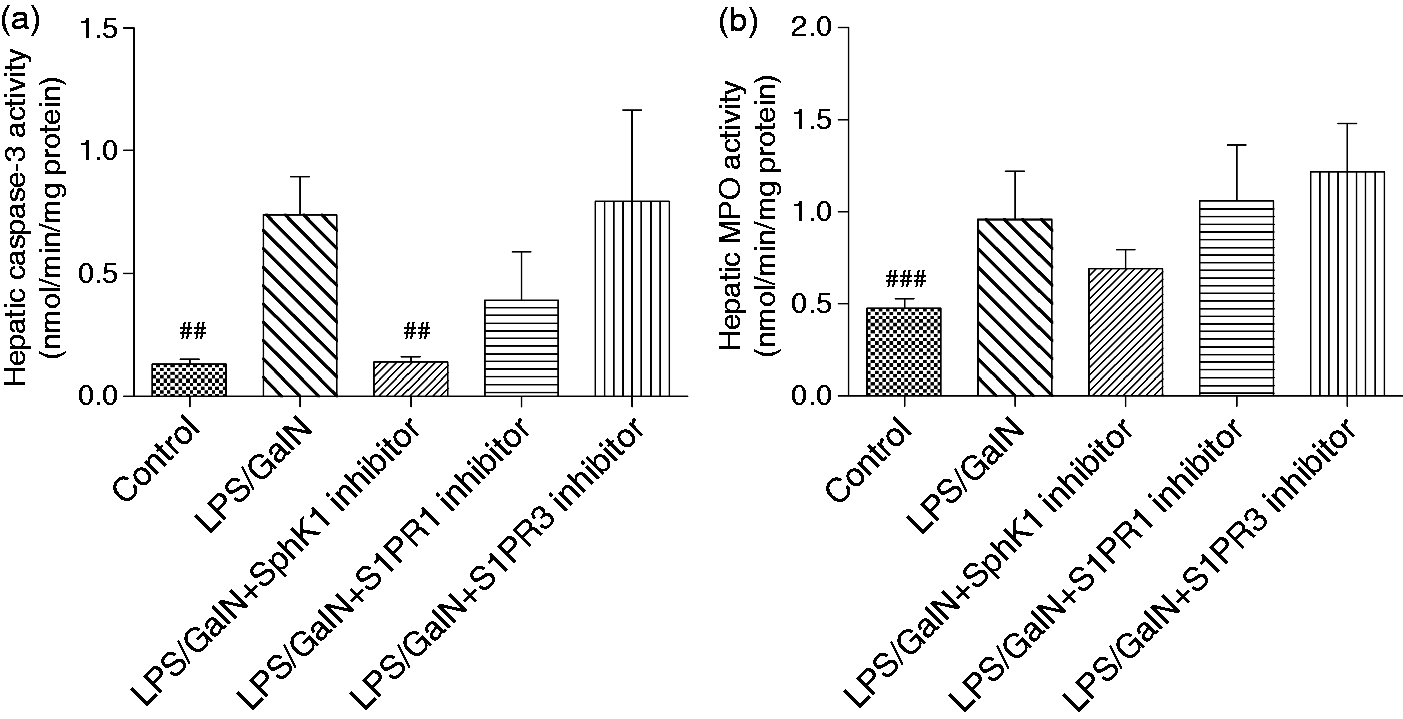
The activation of the MAPKs family was diminished by SphK1 inhibition
MAPKs signaling including JNK, ERK and p38 kinase has been identified as an important pathway in modulating TNF-α. Therefore, we next investigated the effect of SphK1 inhibition on the MAPKs family. Figure 6 shows that p-JNK, p-ERK and p-p38 were enhanced in the LPS/GalN-treated mouse, while the SphK1 inhibitor treatment alleviated the phosphorylation of these molecules. Furthermore, S1PR1 or S1PR3 inhibition might diminish the activation of p38 and JNK (Figure 6(a), (b) and (d)). The phosphorylation of ERK was also reduced by S1PR1 or S1PR3 inhibition, but this was not significant (Figure 6(c)).
The activation of MAPKs signaling is inhibited by SphK1 or S1PRs inhibition. The phosphorylation of ERK, JNK and p38 in the liver were evaluated by Western blotting (a). The relative protein levels of p-p38 (b), p-ERK (c) and p-JNK (d) normalized to GAPDH was presented as fold change of control. The difference between groups was analyzed using one-way ANOVA with Bonferroni post hoc test. Data were expressed as the mean ± SD, n = 3; #p < 0.05, ##p < 0.01 vs LPS/GalN group. MAPKs: mitogen-activated protein kinases; SphK1: sphingosine kinase 1; S1PR1: sphingosine-1-phosphate receptor 1; ERK: extracellular signal-regulated kinase; JNK: c-jun N-terminal kinase; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; ANOVA: analysis of variance.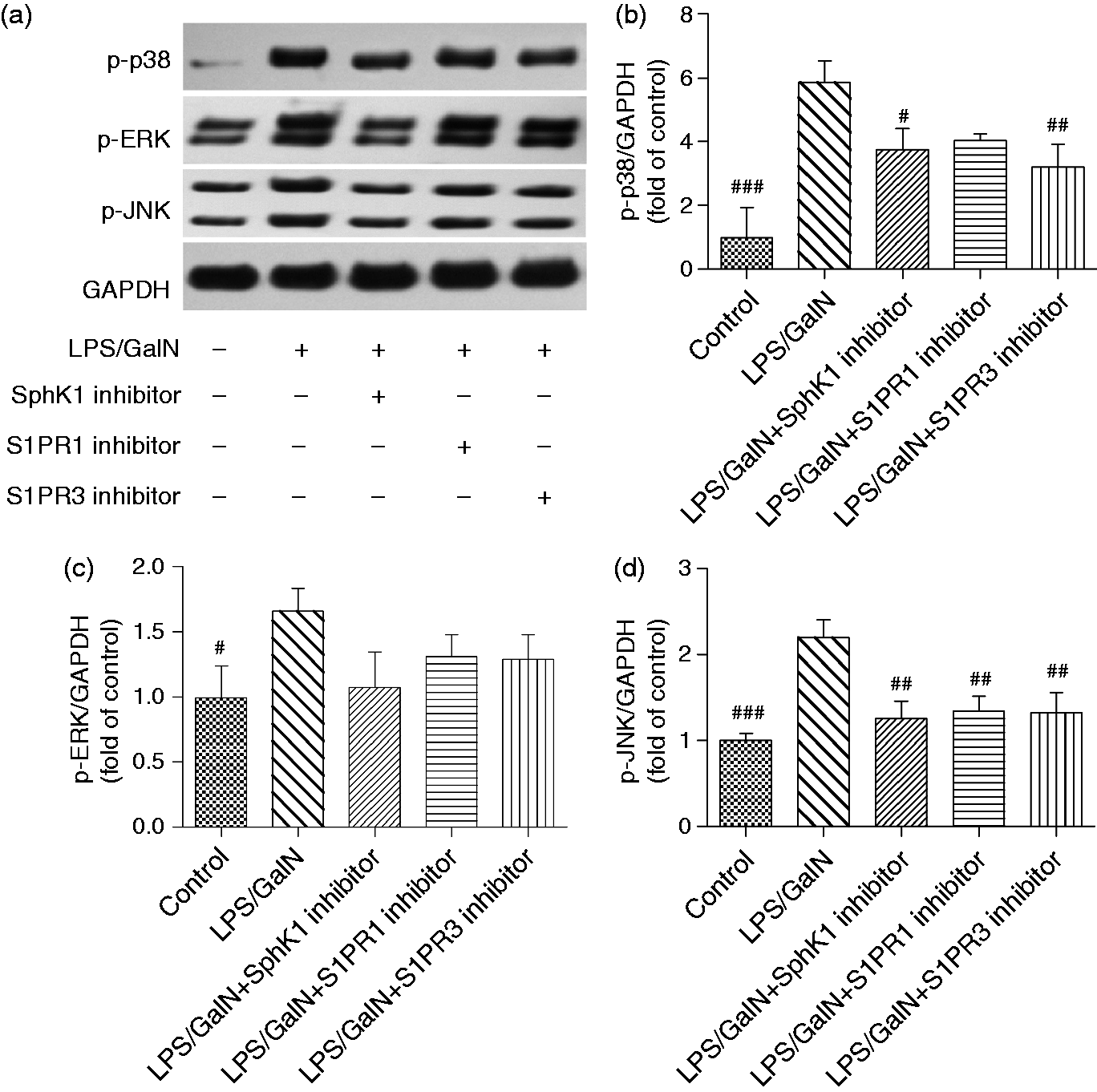
Discussion
In this study, we showed that the expression of SphK1, S1PR1 and S1PR3 was increased in LPS/GalN-treated animals. After SphK1 antagonism, hepatic hemorrhage, serum transaminase activity (AST, ALT), serum TNF-α and IL-6 levels and the activities of hepatic caspase-3 and MPO were remarkably improved. In addition, SphK1 inhibition suppresses the activation of the MAPKs family. The neutralization of S1PR1 or S1PR3 did not improve LPS-induced hepatoxicity, though S1PR1 or S1PR3 inhibition also blocked MAPKs family stimulation.
Recent data have demonstrated that SphK1/S1P/S1PRs signaling is implicated in several inflammatory diseases.6,15,16 SphK1/S1P signaling is involved in the development of severe acute pancreatitis. 6 SphK1-S1PR3 signaling participates in the mechanism of sepsis-associated coagulation and inflammation in dendritic cells. 5 SphK1/S1P/S1PR1 signaling modulates the inflammatory responses related to lung vascular permeability induced by LPS and thrombin. 17 Our results show that LPS/GalN augments the protein level of hepatic SphK1, which is in accordance with previous reports showing that SphK1 expression is upregulated in LPS-mediated inflammation.2,3 The expression of S1PR1 was also facilitated in the LPS/GalN group. Similarly, Kono et al. have found that S1PR1 is activated in LPS-treated hepatocytes. 18 We also showed that S1PR3 protein levels were enhanced in the LPS/GalN group, which is identical to the finding of an elevated expression of S1PR3 in a mouse model of LPS-induced acute lung injury. 19 These results indicate that the SphK1 pathway might be involved in the inflammatory responses associated with LPS/GalN-mediated acute liver failure.
A previous study has concluded that lacking SphK1 attenuates serum cytokines and ameliorates survival rate in a mouse model of LPS-induced sepsis. 5 To further classify the role of SphK1-S1PRs in acute liver failure, we decided to antagonize SphK1 and its receptors, S1PRs, with specific inhibitors. We found that SphK1 inhibition protects against LPS/GalN-mediated liver damage in terms of hepatic hemorrhage, serum transaminase activity and serum TNF-α and IL-6 levels (Figures 2–4). Furthermore, caspase-3, a protease that plays a crucial role in apoptosis, 20 was attenuated by SphK1 inhibition. Hepatic MPO activity, the marker of neutrophil infiltration, 21 was also reduced by the SphK1 inhibitor treatment. These data establish that SphK1/S1P/S1PRs signaling might contribute to the procession of LPS/GalN-induced acute liver failure.
TNF-α is a key mediator in the development of acute liver failure. 22 The induction of TNF-α occurs in the early phase of liver inflammation, which results in the production of several inflammatory cytokines and chemokines. 23 Furthermore, TNF-α initiates the activation of various signaling pathways such as the MAPKs pathway. Several studies have reported that serum TNF-α and IL-6 are dramatically increased in acute liver-failure patients. 24 Neutrophil infiltration, elicited by TNF-α, contributes to liver injury. 25 With regard to cell death, caspase-3 is involved in TNF-α-mediated liver cell death and can be triggered by TNF-α, 23 which may cause hepatocyte apoptosis. In this study, enhanced serum TNF-α and IL-6 levels in response to LPS/GalN were observed in the acute liver-failure animals. However, the neutralization of SphK1 decreased the TNF-α and IL-6 concentrations. SphK1 blockade suppressed the augmented activities of hepatic caspase-3 and MPO triggered by TNF-α. The results indicate that SphK1 blockade might decrease TNF-α induction to alleviate liver damage.
The MAPKs family, including JNK, ERK and p38 kinase, are considered crucial signaling factors involved in modulating TNF-α. 26 These signaling pathways play vital roles in controlling cell death, proliferation, survival and migration. 27 The MAPKs pathway can be stimulated by several environmental stimuli including LPS, which contributes to the production of various inflammatory mediators, such as TNF-α, IL-6 and NO. Das et al. have concluded that the production of TNF-α needs JNK in LPS-induced hepatitis. 28 The neutralization of ERK, p38 or JNK with specific inhibitors attenuates TNF-α expression,29–31 which further emphasizes the importance of the MAPKs in regulating TNF-α. Furthermore, various reports have shown that the MAPKs pathway can be activated in LPS/GalN-induced liver damage.32,33 And TNF-α can initiate the activation of the MAPKs pathway, which results in cell death, gene expression and the production of several pro-inflammatory mediators. 23 The MAPKs pathway has also been regarded as a crucial signaling in acute liver failure. 23 Our study indicates that JNK, ERK and p38 were activated by LPS/GalN, which is consistent with previous reports.8,32 The activated MAPKs pathway leads to the increased production of TNF-α and IL-6 (Figure 4). These data suggest that MAPKs signaling participates in the development of LPS/GalN-induced liver injury. However, we showed that the activation of JNK, ERK and p38 can be blocked by the addition of the SphK1 inhibitor. As the activation of the MAPKs pathway was blocked by SphK1 inhibition, the secretion of inflammatory cytokines such as TNF-α and IL-6 was diminished. 23 The excessive inflammatory responses in the liver may be ameliorated.22,23 As a result, the hepatocyte death and liver neutrophil infiltration are improved. These findings suggest that the protective effect of SphK1 inhibition on LPS/GalN-induced liver damage may be associated with the attenuated activation of MAPKs signaling.
Previous reports have demonstrated that S1PRs are involved in LPS-associated inflammation. 14 As S1PR1 and S1PR3 of S1PRs have been demonstrated to be associated with inflammation, 34 we decided to detect these protein levels. S1PR1 and S1PR3 expression can be upregulated by LPS in human gingival epithelial cells, which promotes the production of inflammatory cytokines. 14 In our study, expression of S1PR1 and S1PR3 were also increased by LPS/GalN (Figure 1(b) and (c)). It has been documented that silencing of S1PR3 by specific small interfering (siRNA) can improve ethanol-associated liver injury via suppressing the phosphorylation of ERK. 35 We also found that S1PR1 or S1PR3 inhibition diminished the phosphorylation of JNK, ERK and p38 (Figure 6). Furthermore, we observed that the inhibition of S1PR1 or S1PR3 decreased the serum levels of TNF-α and IL-6 (Figure 4), which is in accordance with a previous study showing that S1PR3–/– reduces cytokine levels in dendritic cells following LPS challenge. 5 It can be explained that the suppressive activation of JNK, ERK and p38 by S1PR1 or S1PR3 inhibition results in decreased serum TNF-α and IL-6 levels. However, LPS/GalN-mediated liver damage such as hepatic hemorrhage, serum transaminases activity and hepatic caspase-3 and MPO activity was not significantly improved by S1PR1 or S1PR3 inhibition (Figures 2, 3 and 5). The precise role of S1PR1 and S1PR3 in LPS/GalN-induced acute liver failure needs to be further explored.
The main shortcoming of this study is that we neutralized SphK1 and S1PRs only with specific reagents and did not use siRNA or small hairpin RNA (shRNA) to inhibit SphK1 and S1PRs. Experiments overexpressing SphK1 or S1PRs should also be performed.
In summary, we show that SphK1, S1PR1 and S1PR3 are activated by LPS/GalN. SphK1 inhibition may alleviate LPS/GalN-induced liver injury via inhibiting activation of MAPKs signaling. These observations suggest that SphK1-S1PR1,3 signaling play a role in the development of LPS/GalN-induced acute liver failure. SphK1 may be identified as a potential target for the treatment of sepsis-associated liver injury.
Footnotes
Funding
This work was supported by the Medical Science and Technology Innovation Subject of Nanjing Military Region (No. 12Z27).
Conflict of interest
None declared.