
Abstract
Vasculitis comprises a spectrum of disorders defined inflammation of the blood vessel walls and is categorized based on vessel size into small-, medium-, and large-vessel vasculitis. The histological characteristics of leukocytoclastic vasculitis include neutrophil infiltration around small cutaneous blood arteries and fibrinoid necrotizing inflammation. Annular morphology is a unique and rare type of leukocytoclastic vasculitis. We report a case of annular leukocytoclastic vasculitis in a 39-year-old Saudi man who presented with scaly annular erythema and targetoid lesion configurations with prominent, raised borders. He was managed with oral prednisolone 40 mg daily as a tapered dosage regimen, in combination with topical mometasone furoate 0.1% cream applied twice daily, resulting in complete resolution of lesions. To the best of our knowledge, this is the first reported case of annular morphology of leukocytoclastic vasculitis with microvascular occlusions in the Middle East.
Introduction
Vasculitis is a heterogeneous group of conditions marked by inflammation of the walls of blood vessels. According to the 2012 Revised International Chapel Hill Consensus Conference nomenclature, vasculitis is categorized into large-, medium-, and small-vessel vasculitis based on the size of the vessels that are primarily affected, with additional classification based on particular clinicopathologic features. 1 Leukocytoclastic vasculitis (LCV) is histologically defined by fibrinoid necrotizing inflammation and neutrophilic infiltration surrounding small cutaneous blood vessels. Annular morphology is a rare variant of LCV. 2 LCV has a complex etiology, involving both primary and secondary factors playing a role in its pathogenesis. Primary LCV is categorized as idiopathic, whereas secondary LCV can be linked to various conditions, including inflammatory bowel disease, infections (e.g. bacterial or viral), medications (e.g. antibiotics or nonsteroidal anti-inflammatory drugs), autoimmune diseases (e.g. rheumatoid arthritis or systemic lupus erythematosus), cancers (e.g. solid tumors or lymphoproliferative disorders), and systemic vasculitis (e.g. granulomatosis with polyangiitis). Although the precise mechanisms by which these primary and secondary factors cause LCV are not entirely understood, it is thought that immune complex deposition, complement activation, cytokine dysregulation, and endothelial cell injury are important factors in the initiation and continuation of the vasculitic process. Appreciating the heterogeneous causes of LCV is fundamental to precise diagnostic evaluation, optimizing clinical management, and improving patient outcomes.3,4
Case presentation
A 39-year-old Saudi man with a known case of below-knee vessel disease occlusion, paraplegia of the lower limb, and mental retardation was referred from a rehabilitation clinic, complaining of multiple asymptomatic bilateral erythematous plaques over the lower limbs for the past 4 months. He was not taking any medications. His caregiver denied any preceding infection or recent vaccination prior to the onset of the lesions. He was a non-smoker and reported no alcohol consumption. There was no relevant family history of autoimmune, rheumatologic, or dermatologic diseases. All systemic symptoms including fever, arthralgia, abdominal pain, hematuria, or weight loss were negative. Dermatological examination revealed multiple well-defined, non-blanchable, erythematous-to-violaceous scaly maculopapular lesions, and annular-to-polycyclic plaques with an inner collarette of scale. Some lesions exhibited concentric annular (target-like) configurations with prominent, raised borders (Figure 1). Hematological evaluation demonstrated a leukocyte count of 3.77 × 109/L (reference range: 4–10 × 109/L) and elevated monocyte count of 15.4 × 109/L (reference range: 2–10 × 109/L), with a platelet count of 274 × 109/L (reference range: 150–450 × 109/L). Renal function testing revealed low serum creatinine at 45.2 µmol/L (reference range: 62–106 µmol/L). Liver function testing showed elevated alanine aminotransferase at 72.70 U/L (reference range: 10–50 U/L), while all other liver and renal parameters were within normal limits (Table 1). Immunoserological assessment was notable for a positive anti-β2 glycoprotein I immunoglobulin G (anti-β2GPI IgG). All other immune profile screening tests, including hepatitis serology, human immunodeficiency virus-1/2 antigen–antibody combination assay, anti-nuclear antibody, extractable nuclear antigen, complement components C3 and C4, anti-neutrophil cytoplasmic antibodies, anti-La, anti-β2GPI IgG, and anti-cardiolipin IgM and IgG, were negative. A computerized tomography angiogram of the lower extremities revealed a below-knee occlusion involving the mid-popliteal segment, with stenosis/occlusion of the right anterior tibial artery. A skin biopsy extending to the subcutis was performed in December 2024. Histopathologic examination showed minimal spongiosis. The upper dermal small vessels were densely infiltrated by neutrophilic inflammatory cells associated with lymphocytes and macrophages. Perivascular neutrophils displayed degeneration and nuclear dust (karyorrhexis). Fibrinoid necrosis of vessel walls was apparent. There was extravasation of erythrocytes and fibrin as well. There was no dermal mucin deposition. The Periodic acid–Schiff–Diastase special stain did not show basement membrane thickening or fungal micro-organisms. The overall histologic picture was indicative of LCV (Figure 2). Based on these clinical and histopathological findings, a diagnosis of annular leukocytoclastic vasculitis (ALV) was confirmed. A month later, he was treated with oral prednisolone 40 mg daily as a tapered dosage regimen, in combination with topical mometasone furoate 0.1% cream applied twice daily. At the 2-week follow-up, he showed noticeable clinical improvement. By the 4-week follow-up, complete remission was achieved, with only residual post-inflammatory hyperpigmentation. However, a relapse was noted during the subsequent 2-month follow-up visit, for which he was treated with topical clobetasol twice daily for 2 weeks. The antiphospholipid profile was repeated 12 weeks after the first sample, and all serologic tests, including anti-β2GPI IgG, were negative.
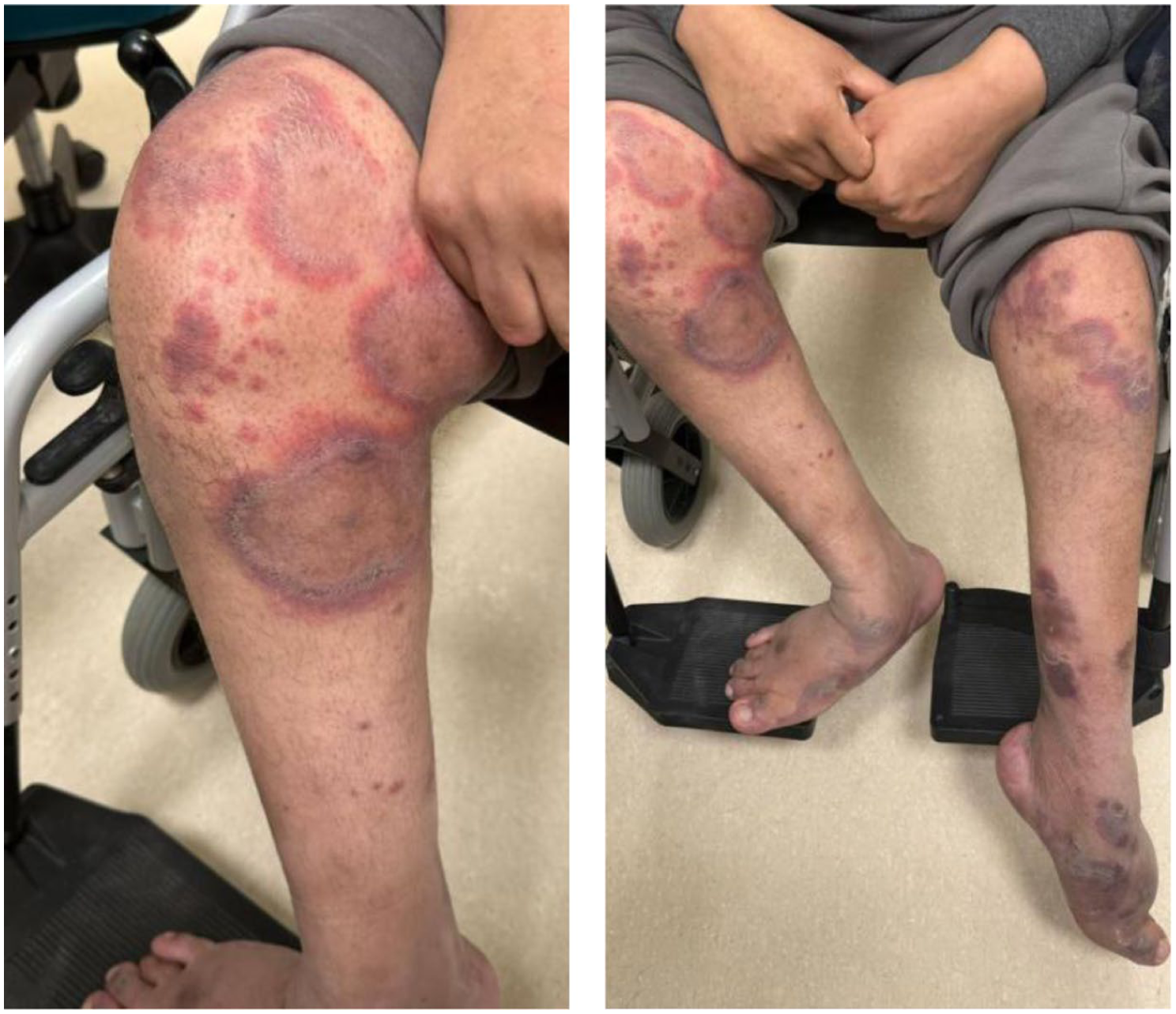
Multiple targetoid lesions with raised borders forming annular and polycyclic plaques on the lower limbs, consistent with the clinical diagnosis of annular leukocytoclastic vasculitis.
Summary of laboratory investigation results.

ALP: alkaline phosphatase; ALT: alanine aminotransferase; AST: aspartate aminotransferase.

(a) Patchy perivascular inflammatory cell infiltrate in the dermis and subcutaneous tissue (H&E stain, ×20 magnification), (b) Dermal vasculature cuffing of inflammatory cells around dermal vessels (H&E stain, ×40 magnification), (c) Fibrinoid necrosis of the vessel wall, fulfilling a key diagnostic criterion of small-vessel vasculitis (H&E stain, ×400 magnification), and (d) Prominent karyorrhectic debris (leukocytoclasia), confirming neutrophilic vessel wall injury characteristic of leukocytoclastic vasculitis (H&E stain, ×400 magnification).
Discussion
ALV is a rare morphological variant of LCV, which is distinguished clinically by annular, polycyclic, or targetoid purpuric plaques rather than the classic palpable purpura. 5 The striking annular and concentric configurations observed in our patient constituted the most distinctive feature of this case and were central to the diagnostic considerations. This morphological presentation is uncommon and could be confused with other inflammatory or infectious skin conditions, such as cellulitis or erythema annulare centrifugum. This circumstance highlights the importance of the clinicopathologic correlation. 1 This case is particularly noteworthy due to the coexistence of significant underlying vascular pathology, specifically below-knee arterial occlusion. Furthermore, chronic vascular insufficiency, stasis, and altered hemodynamics could facilitate local immune complex deposition and endothelial damage, thus increasing the likelihood of cutaneous vasculitic reactions. 6 Nonetheless, in the absence of direct mechanistic evidence, a causal link between vascular occlusion and the onset of ALV remains unproven. Consequently, the observed coexistence in this case should be considered an association rather than evidence of causation. The differential diagnosis for the annular erythematous plaques on the lower limbs encompasses infectious processes like cellulitis, which can manifest with erythema and edema; however, it does not exhibit a non-blanchable purpuric component or histopathologic characteristics of vasculitis. This distinction is clinically significant, as demonstrated by a prior study conducted by Buck et al., which reported that LCV presents similarly to cellulitis in patients with Crohn’s disease, which emphasizes that vasculitis should be considered when lesions are bilateral, purpuric, non-responsive to antibiotics, or recurrent. 7 In this case, the chronic nature, bilateral appearance, annular morphology, and confirmation by histology supported a diagnosis of ALV, rather than infectious cellulitis. Given that no systemic inflammatory, infectious, or hematologic disorder was found, the isolated monocytosis was considered as nonspecific and lacking a clear clinical correlation. 8 A key aspect of this case is the temporary presence of anti-β2GPI IgG antibodies. These antibodies are usually associated with antiphospholipid syndrome (APS) and are known to cause endothelial activation, complement activation, and microvascular injury. 9 The presence of anti-β2GPI IgG in this patient raises the possibility that subclinical endothelial dysfunction or immune-mediated vascular injury may have contributed to the development of ALV. Nevertheless, our patient does not fulfill the revised Sydney diagnostic criteria for the diagnosis of the true APS. According to this criterion, the diagnosis of APS requires at least one clinical criterion, such as arterial or venous thrombosis or defined pregnancy morbidity, and one laboratory criterion, with persistent antiphospholipid antibodies positivity demonstrated on two occasions at least 12 weeks apart. 10 Although our patient had documented below-knee arterial occlusion, anti-β2GPI IgG positivity was transient and reverted to negative on repeat testing after 12 weeks, thereby failing to meet the requirement of laboratory persistence. False-positive antiphospholipid antibody results are recognized, and these antibodies may be detected in up to 12% of the general population, with increasing prevalence with age. In the absence of clinical APS, they may also occur in association with infections, medications, or malignancy. Although central to the pathogenesis of APS-related thrombosis, most individuals with isolated, low-titer, or non-persistent antibodies do not develop the syndrome. 11 Therefore, these findings are best interpreted as isolated, transient anti-β2GPI positivity rather than definitive APS. The histopathological examination confirmed the classic features of LCV, which include perivascular neutrophilic infiltration with leukocytoclasia, fibrinoid necrosis of small vessel walls, and erythrocyte extravasation. These parameters supported the diagnosis of cutaneous small vessel vasculitis and excluded infectious or connective tissue disease-related mimickers, as aligned with the study by Chanprapaph et al. 12 The absence of dermal mucin deposition, interface dermatitis, or fungal elements further strengthened this conclusion. Notably, the patient responded favorably to systemic corticosteroids with complete remission of disease, although relapse occurred, and it is consistent with the relapsing nature of cutaneous LCV when underlying predisposing factors persist. 13 This clinical course highlights the importance of ongoing surveillance and management of contributing vascular or immunological conditions (Supplemental Material).
Conclusion
This case shows a rare annular presentation of LCV, which occurred in the context of transient anti-β2GPI IgG positivity and significant underlying vascular disease. While criteria for APS were not met, this presentation suggests that anti-β2GPI antibodies and chronic vascular insufficiency may play contributory roles in selected cases of cutaneous vasculitis. Awareness of this morphology and its mimickers is essential to avoid misdiagnosis and ensure appropriate management.
Supplemental Material
sj-pdf-1-sco-10.1177_2050313X261440652 – Supplemental material for Annular leukocytoclastic vasculitis associated with isolated anti-β2 glycoprotein I immunoglobulin G positivity: A case report
Supplemental material, sj-pdf-1-sco-10.1177_2050313X261440652 for Annular leukocytoclastic vasculitis associated with isolated anti-β2 glycoprotein I immunoglobulin G positivity: A case report by Saleha Abdulrahman Aldawsari, Ajlan Hashim Alajlani, Abdulelah Nabil Alkadi and Sadeq Wasil Al-Dandan in SAGE Open Medical Case Reports
Footnotes
ORCID iDs
Consent for publication
The authors obtained written consent from the patient for his photographs and medical information to be published in print and online and with the understanding that this information may be publicly available.
Author contributions
Saleha Abdulrahman Aldawsari: idea, picture preparation, manuscript editing, manuscript review. Ajlan Hashim Alajlani and Abdulelah Nabil Alkadi: literature search, manuscript preparation. Sadeq Wasil Al-Dandan: picture preparation.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.