
Abstract
Kikuchi–Fujimoto disease (KFD) is a rare, benign, self-limited lymphadenitis most commonly affecting children and young adults, particularly females of Asian descent. Its etiology is unclear but is thought to involve immune dysregulation triggered by infection. We report a 14-year-old South Asian female presenting with 8 days of high fever, frontal headache, bilateral eye redness, transient arthralgias, and palpable lymphadenopathy in the cervical, axillary, and supraclavicular regions. Her history included recent travel to multiple countries and a prior episode of fever of unknown origin attributed to Bartonella. Initial laboratory evaluation revealed pancytopenia, elevated transaminases, and increased erythrocyte sedimentation rate and lactate dehydrogenase with normal C reactive protein. Infectious workup was negative for malaria, cytomegalovirus, West Nile virus, Rickettsia, Bartonella, and dengue. Ultrasound of the left axilla and computed tomography showed bilateral axillary lymphadenopathy. A cytokine panel demonstrated markedly elevated IL-18. Definitive diagnosis was established by axillary lymph node biopsy, which revealed histiocytic necrotizing lymphadenitis consistent with KFD. Systemic inflammatory disorders such as systemic-onset juvenile idiopathic arthritis, multisystem inflammatory syndrome in children, and Kawasaki disease were considered and excluded based on clinical, laboratory, and histopathological findings. The patient’s fever resolved after dexamethasone administration, supporting an immune-mediated process. She subsequently developed recurrent fever, rash, and oral ulcers, which responded rapidly to intravenous corticosteroids, followed by a steroid taper.
Teaching points
KFD should be considered in children and adolescents with persistent fever, lymphadenopathy, and cytopenia, especially in those of Asian descent.
The differential diagnosis is broad and includes infectious, malignant, and systemic inflammatory disorders; histopathological confirmation is essential.
Corticosteroids may be effective in severe or refractory cases.
Early recognition and multidisciplinary evaluation are critical to avoid misdiagnosis and unnecessary interventions.
Educational objectives
To enhance understanding of Kikuchi–Fujimoto disease (KFD) by exploring its clinical presentation, including prolonged fever, lymphadenopathy, and cytopenia, and to emphasize the importance of considering KFD in the differential diagnosis of similar symptom profiles to prevent misdiagnosis.
To explain the role of interferons in autoimmune diseases, particularly in systemic lupus erythematosus (SLE), and its impact on the clinical management of patients with overlapping features of KFD and SLE.
Introduction
Kikuchi–Fujimoto Disease (KFD) is a rare benign condition associated with fever, fatigue, and lymphadenopathy, especially in cervical and postauricular areas.1 –6 The cause of this disease remains elusive, but it is postulated to be triggered by recent infection.1 –3 In this report, we discuss a case of a teenager who presented with prolonged fever of unknown etiology, complicated by confounding exposure histories and lab markers. Her case was puzzling to many physicians because she not only had fever but also joint pain, cytopenia, and lymphadenopathy in the postauricular and axillary areas. In addition, she had a prior history of hospitalization for fever of unknown origin, ultimately found to be secondary to Bartonella, and had an extensive travel to Kenya and India. These factors warranted an extremely comprehensive workup involving multiple subspecialties and an invasive procedure for biopsies. 7 KFD continues to be a strenuous diagnosis, requiring attentiveness and broad differentials. This report aims to discuss a constellation of symptoms that could be seen in a patient with KFD to avoid misdiagnosis in this patient population.
Case
Hospital course
A 14-year-old South Asian female was admitted with an 8-day history of high fever (Tmax 104°F), frontal headache, and bilateral eye redness without discharge. She reported transient arthralgias in her elbows and wrists, which resolved prior to admission, and managed her fever with acetaminophen and ibuprofen. She had a recent positive streptococcal swab and received a dose of Bicillin at urgent care. During hospitalization, she experienced intermittent emesis but denied diarrhea or respiratory symptoms.
Her travel history included recent travel to San Ramon, California, and prior trips to the United Kingdom, France, Kenya, and India, raising initial concern for infectious etiologies such as malaria, dengue, and Bartonella. Family history was negative for malignancy or autoinflammatory disorders.
Initial labs revealed pancytopenia, anemia, thrombocytopenia, and elevated transaminases. erythrocyte sedimentation rate (ESR) and lactate dehydrogenase were also elevated, while C reactive protein (CRP) was normal (Table 1). Physical exam showed palpable lymphadenopathy in the cervical, axillary, and supraclavicular regions without joint swelling. Hematology, rheumatology, oncology, and interventional radiology were consulted. Infectious workup was negative for malaria, cytomegalovirus (CMV), West Nile virus, Rickettsia, Bartonella, Human Herpesvirus 6 (HHV-6), Coccidioides, and dengue. Mycoplasma pneumoniae immunoglobulin G (IgG) and Epstein Barr Virus (EBV) IgG were positive, indicating past exposure. Complement levels were normal, and a cytokine panel showed markedly elevated IL-18. Imaging with computed tomography (CT) and ultrasound revealed bilateral axillary lymphadenopathy (Figures 1 and 2); echocardiogram was unremarkable.
Consolidated laboratory, diagnostic, and pathology findings.

CMV: Cytomegalovirus; IgG: immunoglobulin G; IgM: immunoglobulin M; CT: computed tomography; PCR: polymerase chain reaction; ELISA: enzyme-linked immunosorbent assay; HHV-6: Human Herpesvirus 6; LN: lymph node; IHC biopsy: immunohistochemistry biopsy.

Ultrasound of left lymph node.

CT chest with left lymph node.
On hospital day four, axillary lymph node and bone marrow biopsies were performed. The patient became afebrile on day five, likely due to dexamethasone administered for airway edema during anesthesia. She was discharged on naproxen with pending biopsy results and follow-up with infectious disease and rheumatology.
After discharge, she developed recurrent fevers, a generalized maculopapular rash on arms and trunk, and oral ulcers (Figure 3). She was readmitted with concern for Stevens–Johnson syndrome secondary to naproxen. However, lymph node biopsy confirmed histiocytic necrotizing lymphadenitis, also known as KFD (Figure 4) and her symptoms were attributed to evolving KFD rather than a drug reaction. IV steroids led to rapid resolution of fever, rash, and oral ulcers, and she was discharged on a 2-week oral steroid taper.

Scattered maculopapular nonpruritic rash on face, arms, and trunk (a), (b), (c). Erythema/early ulcers on upper and lower inner sulci and under tongue, no palate ulcers (e), (f).
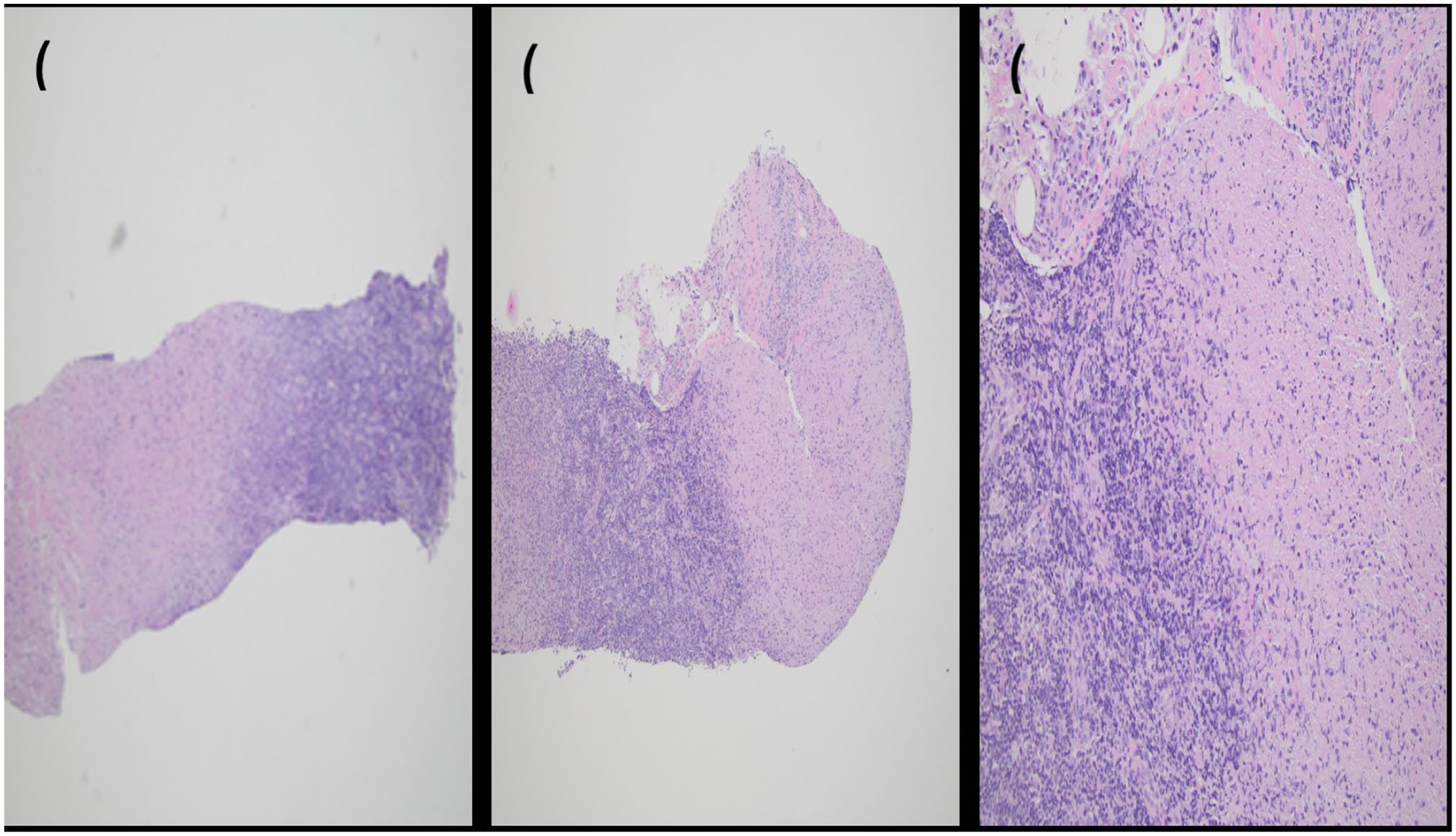
Histopathology of left axillary lymph node core biopsy demonstrating necrotizing lymphadenitis with geographic areas of necrosis, scattered histiocytes, and prominent karyorrhexis, without significant acute inflammatory infiltrate.
Discussion
KFD, or histiocytic necrotizing lymphadenitis, is a rare, benign, and self-limited condition characterized by fever and lymphadenopathy, most commonly affecting children and young adults, with a female predominance and higher incidence in Asian populations.1 –4 The etiology remains unclear but is postulated to involve viral triggers or autoimmune mechanisms.5,6
This 14-year-old South Asian female’s presentation, includes prolonged fever, lymphadenopathy, cytopenias, and rash, was consistent with KFD, which typically affects young women of Asian descent (see Table 3 for timeline of events).1 –5 The differential diagnosis was broad, given her travel history and initial concern for infectious etiologies. Infections such as malaria, CMV, West Nile virus, Rickettsia, and Bartonella can present with overlapping features, including fever, lymphadenopathy, cytopenias, and transaminitis.8,9 These were systematically excluded through targeted testing (see Tables 1, 2 for laboratory findings and Figure 4 for histology results). Below is a list of shared features between these infections and KFD:
Positive IgG titers for EBV and Mycoplasma indicated past infections without current implications. The presence of lymphadenopathy and cytopenia also raised concern for malignancy, particularly lymphoma, but imaging and laboratory findings were not supportive.
Hematologic and immunologic workup.

ALP: Alkaline phosphatase; AST: Aspartate aminotransferase; ALT: Alanine aminotransferase; LDH: Lactate dehydrogenase; ESR: Erythrocyte sedimentation rate; CRP: C reactive protein; ANA: Antinuclear antibody; ASO: Antistreptolysin O.
Timeline of events.

LDH: Lactate dehydrogenase; ESR: Erythrocyte sedimentation rate; SLE: Systemic lupus erythematosus; IgG: immunoglobulin G; CRP: C reactive protein; CMV: Cytomegalovirus; KFD: Kikuchi-Fujimoto Disease; VUS: variant of uncertainsignificance; EBV: Epstein-Barr Virus; HHV-6: Human Herpesvirus 6.
In addition to infectious and malignant etiologies, several systemic inflammatory disorders can present with fever, lymphadenopathy, rash, and laboratory evidence of systemic inflammation in children. Notably, systemic-onset juvenile idiopathic arthritis (sJIA, Still’s disease), multisystem inflammatory syndrome in children (MIS-C), and Kawasaki disease were considered in this case.10 –13
Systemic-onset juvenile idiopathic arthritis (sJIA/Still’s disease) is characterized by quotidian fever, evanescent salmon-pink rash, arthritis, lymphadenopathy, hepatosplenomegaly, and serositis. While our patient had fever, rash, and lymphadenopathy, she lacked persistent arthritis, hepatosplenomegaly, and serositis. Laboratory findings in sJIA often include markedly elevated ferritin, CRP, and ESR, with possible leukocytosis and thrombocytosis, whereas our patient had pancytopenia and normal CRP. The absence of arthritis and the histopathological findings on lymph node biopsy further argued against sJIA.10,11
MIS-C is a postinfectious complication of SARS-CoV-2, presenting with persistent fever, mucocutaneous involvement, gastrointestinal symptoms, lymphadenopathy, and elevated inflammatory markers. Our patient did not have a history of recent COVID-19 infection or exposure, and her CRP was normal. She also lacked significant gastrointestinal symptoms, hypotension, or cardiac dysfunction on echocardiogram, making MIS-C less likely. 12
Kawasaki disease typically presents with prolonged fever, conjunctival injection, mucous membrane changes, rash, extremity changes, and cervical lymphadenopathy. While our patient had fever, eye redness, rash, and oral ulcers, she did not meet the full clinical criteria for Kawasaki disease, and echocardiogram was unremarkable. Additionally, the age and ethnicity were less typical, and the lymph node biopsy was not consistent with Kawasaki disease. 13
The exclusion of these systemic inflammatory disorders was based on the clinical presentation, laboratory findings, imaging, and, most definitively, the histopathological diagnosis of KFD.
The patient’s fever resolution after anesthesia was likely due to dexamethasone, which has been shown to be effective in managing severe KFD symptoms, including fever and lymphadenopathy.14 –17 The rapid resolution of fever following dexamethasone administration is notable. Dexamethasone, a potent corticosteroid, exerts broad anti-inflammatory and immunosuppressive effects. The patient’s prompt defervescence after a single dose suggests that her fever was driven by an underlying inflammatory or immune-mediated process, rather than a persistent infectious etiology. This clinical response further supports the diagnosis of KFD, which is characterized by immune dysregulation and heightened cytokine activity. KFD is generally self-limited, with most cases resolving within 1–4 months.1,2,5 Corticosteroids are reserved for severe or refractory cases and have demonstrated efficacy in controlling symptoms.14 –17 There is a rare but recognized association between KFD and systemic lupus erythematosus, warranting ongoing monitoring for the development of autoimmune features. 7
Genetic testing revealed variants of uncertain significance, including TMEM173, associated with interferonopathies such as STING-Associated Vasculopathy with Onset in Infancy. The patient’s cytokine panel showed elevated IL-18 and interferon gamma, consistent with active interferon signaling. 5 Given her current stability, ongoing clinical monitoring is recommended, with consideration of further interferon signature testing if symptoms recur.
Final diagnosis
KFD is notoriously challenging to diagnose due to its nonspecific symptoms and the necessity for histopathological confirmation. In this case, the diagnosis was confirmed through an axillary lymph node biopsy, which revealed histiocytic necrotizing lymphadenitis, consistent with KFD. The patient’s clinical presentation, including fever, lymphadenopathy, and subsequent development of generalized rash and oral ulcers following naproxen administration, aligns with reported manifestations of KFD. 2
Conclusion
KFD is a rare, self-limited condition characterized by fever, fatigue, and lymphadenopathy, commonly in the cervical or postauricular regions. The involvement of multiple specialties underscores the complexity of KFD diagnosis. This multidisciplinary approach facilitated the exclusion of other potential diagnoses, such as lymphoma, autoimmune disorders, and other infectious etiologies. KFD, though rare, should be considered in patients with persistent fever and lymphadenopathy, especially when infectious and autoimmune screenings are inconclusive. The elevated IL-18 levels support the inflammatory nature of KFD, and the response to steroids highlights their role in managing this condition. Clinicians should keep KFD in mind when evaluating fever and cytopenia in young women, even when infectious history is prominent. Early recognition and appropriate histopathological examination are crucial in preventing misdiagnosis and ensuring timely, effective treatment.
Footnotes
Acknowledgements
We would like to acknowledge the support of CHOC hospital for providing the necessary resources and facilities for this study.
Author note
This statement aligns with the guidelines provided in the sources, ensuring clarity and transparency.
Ethical considerations
Our institution does not require ethical approval for reporting individual cases or case series.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Consent to participate
Written informed consent was obtained from the patient and their parents for the publication of their anonymized information in this article.