
Abstract
Granulomatosis with polyangiitis is a rare autoimmune disorder. Granulomatosis with polyangiitis’s signs and symptoms reflect affected organs, which are inclined toward the respiratory tract and kidneys. Gastrointestinal involvement is uncommon and barely represents this disease, particularly as an initial presentation. Here, we describe a diagnostically challenging case of a 14-year-old boy whose first manifestation of granulomatosis with polyangiitis was gastrointestinal bleeding, initially resembling immunoglobulin A vasculitis. Despite overlapping features such as arthralgia and skin lesions, renal biopsy, and cytoplasmic antineutrophil cytoplasmic antibodies positivity distinctly established granulomatosis with polyangiitis diagnosis. Recognizing gastrointestinal manifestations as potential indicators of granulomatosis with polyangiitis, even when hallmark respiratory symptoms are absent, enhances vigilance, and is crucial for early diagnosis and appropriate management.
Introduction
Granulomatosis with polyangiitis (GPA), formerly known as Wegener’s granulomatosis, is a necrotizing vasculitis characterized by inflammation of the vascular wall and perivascular and extravascular granulomatous inflammation. Clinically, GPA is characterized in its full form by ear, nose, and throat (ENT) signs and lung and kidney involvement. 1 However, there might be other organs involved, including the eyes, skin, joints, and nervous system. The gastrointestinal (GI) system manifestations are estimated to occur in only 0%–7% of patients 2 ; therefore, GI bleeding (GIB) is a rare indicator of this disease, especially in children who have a lower incidence of GPA compared to adults. 3 Here, we report a pediatric case initially misdiagnosed as immunoglobulin A vasculitis (IgAv, formerly known as Henoch–Schonlein purpura), whose first manifestation of GPA was GIB. Previous case reports have been in adults, and our patient is a teenage boy. This case report aims to highlight the significance of this issue to prevent potential misdiagnosis or delayed diagnosis in future cases.
Case presentation
A 14-year-old boy was admitted to the rheumatology ward of Imam Hossein Children’s Hospital, Isfahan University of Medical Sciences, presenting with a history of an acute onset of petechiae and purpura lesions on his lower extremities, accompanied by arthralgia, joint swelling in the lower limbs, as well as severe abdominal pain and vomiting.
On the physical examination, the presence of palpable petechiae and purpura was distributed across the lower limbs and buttocks. Also, severe arthritis was affecting both ankles and knees, and significant pitting edema in the lower limbs was notable. Abdominal palpation demonstrated generalized tenderness.
The patient had normal blood pressure and stable vital signs. Laboratory investigations were as complete blood count (CBC), including white blood count (WBC): 14,000/mm3, neutrophil: 75%, platelet (Plt): 335,000/mm3, and hemoglobin (Hb) level of 11 g/dL. Inflammatory markers were mildly elevated, with an erythrocyte sedimentation rate (ESR) of 25 mm/h and C-reactive protein (CRP) of 14 mg/L, which demonstrated that leukocytosis, anemia, and inflammation were present.
Abdominal ultrasonography identified trace-free fluid in the peritoneal cavity, while liver function tests and renal function markers (blood urea nitrogen (BUN) and creatinine (Cr) were normal); urine analysis (U/A) did not indicate hematuria nor proteinuria, thus making hepatic and renal involvement unlikely.
Based on clinical and laboratory findings, a provisional diagnosis of IgAv based on American College of Rheumatology (ACR) criteria 4 was established (Table 1).
ACR criteria: a patient must have at least two of the following four criteria present to be diagnosed with IgAv.

ACR: American College of Rheumatology; IgAv: immunoglobulin A vasculitis.
The patient received supportive care, including hydration and analgesics (Acetaminophen), and was discharged after a 3-day hospitalization with a resolution of acute symptoms.
One week following discharge, he had an acute exacerbation, including abdominal pain with hematochezia, resulting in his readmission. Laboratory tests were repeated and demonstrated as below:
Leukocytosis (WBC: 16,000/mm3; neutrophils: 80%), thrombocytosis (Plt: 480,000/mm3), and anemia (Hb: 10 g/dL). Inflammatory markers were elevated, with an ESR of 45 mm/h and a CRP of 9 mg/L. Renal function remained within normal limits (BUN: 12 mg/dL; Cr: 0.7 mg/dL). U/A revealed microscopic hematuria (5–10 red blood cells/high-power field (hpf)) and mild proteinuria (1+).
He was treated with intravenous pantoprazole and subcutaneous octreotide with the diagnosis of acute GIB. Once his condition was stable, he had an endoscopy and colonoscopy (Figure 1). After procedures, he was discharged with oral proton-pump inhibitors.

Endoscopy showing erosive gastropathy and duodenopathy causing GIB.
Once again, 7 days later, he experienced a flare-up episode with skin lesions, abdominal pain, and severe arthritis affecting the lower extremities (Figure 2). Vital signs remained stable, with normotensive blood pressure.
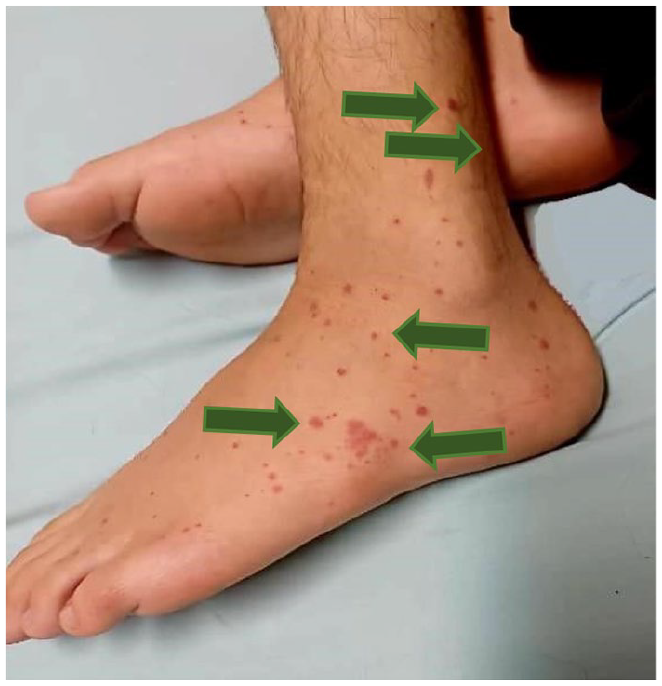
Palpable purpuric lesions in the right leg indicating IgAv.
On physical examination, petechiae and purpura in the mentioned regions were inspected.
Laboratory studies indicated leukocytosis with neutrophilic predominance, marked thrombocytosis (Plt count of 550,000/mm3), and significantly elevated ESR (75 mm/h). Renal parameters showed mild azotemia (BUN: 25 mg/dL; Cr: 0.9 mg/dL). His U/A revealed gross hematuria (30–40 dysmorphic red blood cells/hpf, 40% dysmorphic), heavy proteinuria (3+), and 24-h urinary protein excretion of 954 mg, hence the renal involvement.
In light of new findings and disease progression, including cutaneous, GI, articular, and renal manifestations, the differential diagnoses were expanded to encompass complicated IgA vasculitis or new vasculitis, and therefore, rheumatologic tests were requested to refine the diagnosis.
Serological testing showed negative reactivity for antinuclear antibodies, perinuclear antineutrophil cytoplasmic antibodies, and double-stranded DNA; however, the cytoplasmic antineutrophil cytoplasmic antibodies (c-ANCA) were measured at a level of 850 ng/dL (N < 100 ng/dL).
Serological assays for infectious diseases such as COVID, parvovirus B19, cytomegalovirus, Epstein–Barr virus, and Brucellosis were all negative.
A skin biopsy was performed on the patient, and histopathological analysis of the biopsy identified leukocytoclastic vasculitis, characterized by leukocyte infiltration with the absence of complement and Ig depositions, including IgM, IgG, and IgA, ruling out the IgAv.
Given the c-ANCA seropositivity alongside renal involvement, GPA was suspected. Consequently, a computed tomography scan of the chest and paranasal sinuses was performed, which showed no significant changes. Renal biopsy findings confirmed focal segmental glomerulosclerosis (FSGS) without Ig (IgA, IgM, and IgG) or complement deposition.
Based on the ACR/European Alliance of Associations for Rheumatology (EULAR) diagnostic criteria, 5 the diagnosis of GPA was established (Table 2).
ACR/EULAR diagnostic criteria; sum the scores for 10 items, if present. A score of ⩾5 is needed for the classification of GPA.

ACR: American College of Rheumatology; c-ANCA: cytoplasmic antineutrophil cytoplasmic antibodies; EULAR: European Alliance of Associations for Rheumatology; GPA: granulomatosis with polyangiitis; MPO: myeloperoxidase; p-ANCA: perinuclear antineutrophil cytoplasmic antibodies; PR3: proteinase 3.
A treatment regimen was initiated with intravenous Prednisolone pulse (30 mg/kg), oral Methotrexate, and Cyclophosphamide pulse infusion (500 mg/m2) for 3 continuous days. Following induction therapy, oral prednisolone (2 mg/kg/day) was continued. Cyclophosphamide was administered biweekly for three cycles, followed by triweekly intervals.
After 2 months of therapy, his clinical condition improved a lot as laboratory data illuminates (Table 3), with complete resolution of cutaneous manifestations. Follow-up assessments were as follows: CBC (WBC: 12,800/mm3; neutrophil 75%), normal range inflammatory markers (ESR: 17 mm/h; CRP: 2 mg/L). As for U/A: RBC: 5–10/hpf, protein (1+), and 24-h urine protein: 720 mg. Also, Serum c-ANCA titers declined to 350 ng/dL (N < 100 ng/dL).
Laboratory tests of the first admission, second admission after 1 week, third admission after 2 weeks, and follow-up data after 2 months.
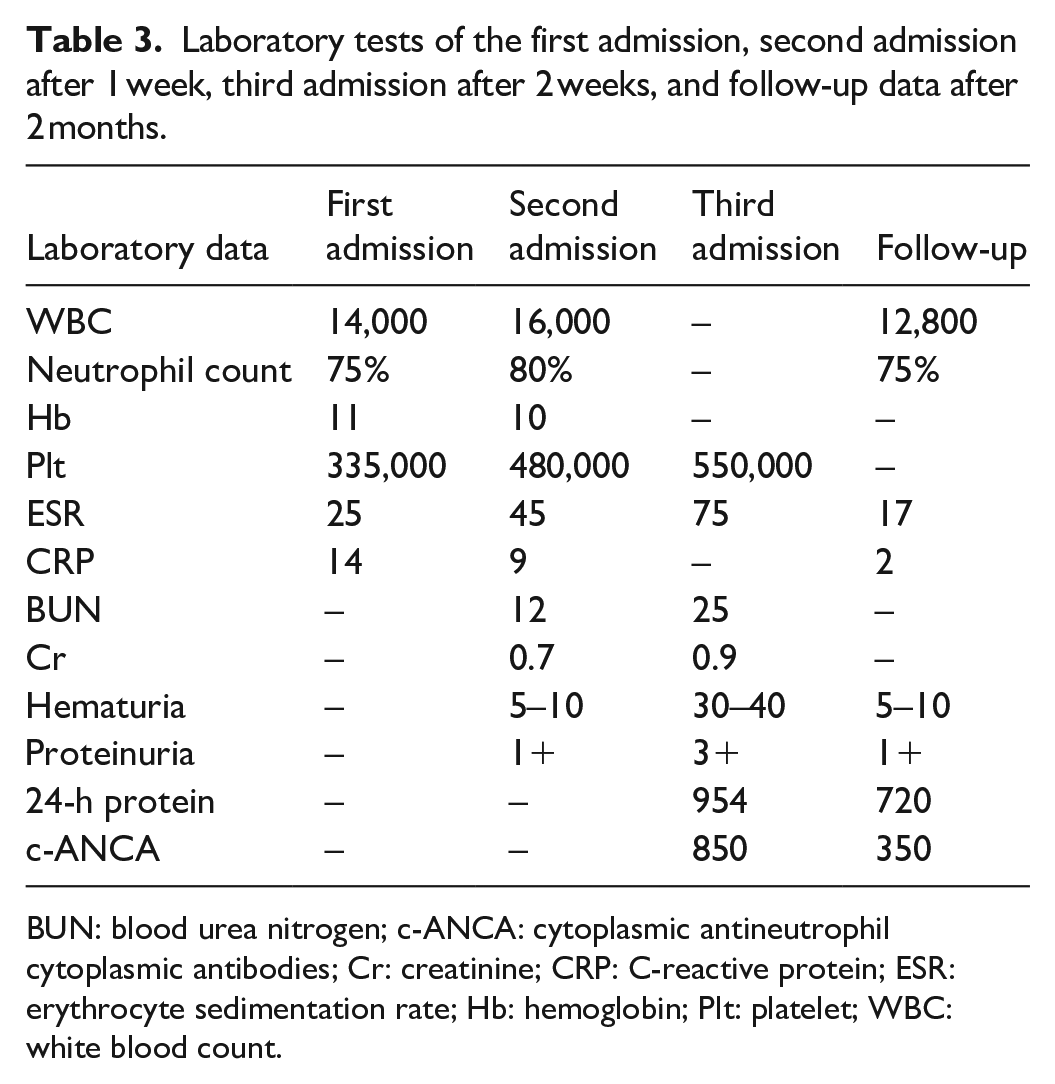
BUN: blood urea nitrogen; c-ANCA: cytoplasmic antineutrophil cytoplasmic antibodies; Cr: creatinine; CRP: C-reactive protein; ESR: erythrocyte sedimentation rate; Hb: hemoglobin; Plt: platelet; WBC: white blood count.
Treatment continued with the goal of maintaining long-term immunosuppression as the ACR/EULAR guidelines suggest, using oral prednisolone, methotrexate, and cyclophosphamide to prevent disease relapse.
Discussion
IgAv represents the most prevalent form of vasculitis in the pediatric population. It typically manifests with palpable purpura, abdominal pain and GIB, renal involvement, and musculoskeletal complaints such as arthralgia or arthritis. 6 In this case, based on initial symptoms such as skin rash, abdominal pain, GIB, and arthralgia, IgA vasculitis was first suspected. However, several key findings as an example, the absence of IgA deposition on skin biopsy, rising c-ANCA titers, and renal progression (proteinuria, hematuria) with histological confirmation via kidney biopsy (FSGS), deviated diagnosis of IgA vasculitis. As there is a clinical overlap of this disease with GPA. 7
GPA is an uncommon condition in the pediatric population. Panupattanapong et al. reported a prevalence of only 3.6% of patients having a childhood onset. 8 The disease more commonly presents in the second to fourth decades of life. A review by Ledó and Pethő documented several cases of GI-dominant GPA, but all patients were adolescents, including an 18-year-old male. 9 However, our patients were significantly younger than previously reported. This highlights that initial GI presentations are almost exclusively reported in older individuals, not in children below the teenage years.
GI involvement is also infrequent, with vomiting and GIB each occurring in ~6% of cases, and bloody diarrhea in 3%. 10 In pediatric subglottic stenosis, ENT involvement and renal findings are often more prominent than GI symptoms. 11
This age discrepancy underscores the clinical significance of our report and highlights awareness in the face of unexpected GIB in young patients, where typical ENT or pulmonary symptoms are absent. These findings furthermore emphasize the exceptional nature of this case.
Conclusion
The initial signs and symptoms of IgAv and prodrome presentation of other vasculitis might be similar. As these two diseases overlap, subsequent diagnosis should be based on more definitive tests, such as IgA deposition on skin for IgAv and C-ANCA for GPA, to differentiate them, especially when there is a discrepancy between treatment and clinical outcome.
Footnotes
Acknowledgements
The authors are thankful to the patient who agreed to publish his case report.
Consent to participate
The written informed consent form was obtained from the patient’s mother to use the information provided for this report.
Consent for publication
We restate that institutional approval is not required to publish the case details.
Author contributions
M.J. gave the conception of the work. H.A.M. and N.C. have drafted the work.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
The data used to support the findings of this study are available from the author upon request.