
Abstract
Type I plasminogen (PLG I) deficiency is a genetic disorder inherited in an autosomal recessive mode and carries high mortality and morbidity. This case report discusses two babies, aged 2 and 3 months, who were diagnosed with ligneous conjunctivitis and congenital hydrocephalus. They had progressive macrocephaly, which led to the insertion of a ventriculoperitoneal shunt. However, there was no significant improvement. During the course of the disease, they underwent genetic testing and were diagnosed with PLG I deficiency. One of the babies underwent ventriculocholecystic shunt insertion as part of palliative care and management, since this disease has poor absorption in the peritoneal cavity. Unfortunately, there was no improvement observed, and he died at 18 months. The other baby received intravenous plasma (10 ml/kg) three times a week, plus using several eye drops daily, with moderate improvement. Promising results are expected with the approved plasminogen, human-tvmh, by the Food and Drug Administration. However, access to the newly approved drug in developing countries is challenging, often hindered by cost or supply issues, necessitating the use of alternative treatments.
Keywords
Introduction
Plasminogen (PLG) deficiency can be acquired or congenital. There are two types of congenital PLG deficiency which are type I hypoplasminogenemia and type II dysplasmincogenemia. 1 Type I plasminogen (PLG I) deficiency is a genetic disorder inherited in an autosomal recessive mode that affects 1.6 per 1 million people, 2 with a female to male ratio 1.55:1. 3 K19E mutation is the most common molecular genetic defect worldwide. 4 This disease is characterized by very low or undetectable PLG level and activity. Therefore, fibrin accumulates at the inflammation site and causes healing delay. 4 The severe form of PLG I deficiency has broad clinical manifestation including ligneous conjunctivitis (LC), ligneous gingivitis, respiratory tract, and female genital tract involvement as well as congenital occlusive hydrocephalus.1,5 The prognosis is variable based on the extent, length, and site of the symptoms. 6
In this case report, we describe two babies who had congenital hydrocephalus related to PLG I deficiency and review the literature to explore its potential pathogenesis, natural course, diagnosis, and treatment.
Case presentations
Case 1
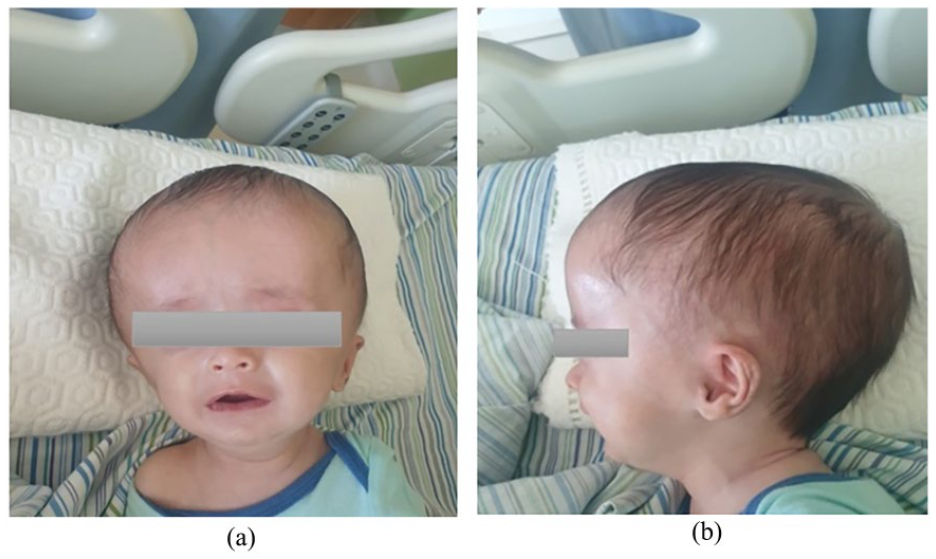
A 2-month-old male patient was admitted to our hospital as a case of increasing congenital hydrocephalus due to malfunctioning ventriculoperitoneal (VP) shunt. Antenatal care started at 6 weeks’ gestational age but was followed by poor visits. At 20 + 3 weeks’ gestation, transabdominal ultrasound showed an abnormally dilated head circumference (HC). Consequently, the mother was referred to do detailed ultrasound, which demonstrated an increase in both biparietal diameter and HC, severe bilateral ventriculomegaly (⩾15 mm), dilated cisterna magna, vermian agenesis, and a dilated third ventricle, all consistent with Dandy–Walker malformation.
At 35 + 3 weeks’ gestation, he was delivered by cesarean section due to high risk of cephalopelvic disproportion with 50 cm HC (greater than 97th percentile) to a mother who is G2P1A0. The baby was admitted to neonatal intensive care unit, 6 days postdelivery, a computerized tomography (CT) scan showed severe hydrocephalus with minimal brain tissue. Afterward, a right occipito-parietal VP shunt was inserted. Three weeks later, he was discharged home with 43.5 cm HC. Postdischarge, the patient continued to develop an increasing HC, associated with recurrent bilateral swelling of the upper and lower eyelids, conjunctival redness, and white lesions. As a result, the patient was referred to an ophthalmologist and diagnosed with LC (Figure 1).
(a, b) Coronal and sagittal views shows hydrocephalus.
At 2 months old, he presented to our neurosurgery clinic for evaluation with HC of 53 cm, alongside a slow filling valve of the shunt. The patient underwent brain magnetic resonance imaging (MRI) without contrast, which showed hugely dilated ventricles and a right shunt catheter in the right posterior horn of the lateral ventricle with minimal brain tissue, and no dilated cisterna magna or vermian agenesis (Figure 2).

(a, b) Sagittal and axial MRI show severe hydrocephalus (red double arrows) with minimal brain tissue (black arrow) and hugely dilated ventricles and right shunt catheter in right posterior horn of lateral ventricle (white arrow).
After assessment, the patient was admitted for observation, shunt revision, and insertion. Family history revealed a third cousin degree of consanguineous parents and a case of the same condition without genetic study confirmation. Therefore, clinical suspicion pointed toward PLG I deficiency, which presents with congenital hydrocephalus and bilateral LC. The patient’s operation was postponed, and upon discharge, he was recommended to follow up with a geneticist. Genomic amplification of exon 14 of the PLG gene and direct sequencing on the DNA extracted from a peripheral blood sample identified c.1752-1753insG (P.cys585valfsx15) mutation in a homozygous manner, confirming the diagnosis of PLG I deficiency. Furthermore, the patient’s parents were advised to undergo genetic and prenatal testing for this gene in the future pregnancies.
Previous two VP shunts were performed before confirming the diagnosis, as the patient’s HC progressively increased to 72 cm due to shunt malfunction. According to literature review, this disease carries high mortality and morbidity. Consequently, the patient was recommended to undergo ventriculocholecystic shunt, as PLG I prevents absorption of cerebrospinal fluid (CSF) from the peritoneal cavity.7,8 Despite ventriculocholecystic shunt insertion at 4 months old, which initially decreased the HC to 62 cm, it subsequently increased back to 73 cm due to shunt malfunction within 1 month. Afterward, multiple VP shunts were inserted, but each resulted in poor outcomes due to thrombotic occlusion, and the patient died at 18 months old.
Case 2
A 3-month-old female patient presented to the hospital by her family complaining of increasing head size and abnormal shape with massive bilateral swelling and hard lesions of both eyelids. At the 14th week of gestation, dilated ventricles were observed. The patient was delivered by cesarean section at 38 weeks of gestation with HC of 37 cm. At one and a half months old, her HC increased progressively by 8.5 cm. A brain CT scan without contrast showed a hugely dilated ventricular system sparing the fourth ventricle, with agenesis of corpus callosum. Because of progressive hydrocephalus, implantation of a VP shunt was recommended. On physical examination, the patient had a wide anterior fontanel measuring about 4 cm in its anteroposterior and 4 cm in its transverse diameter, and a wide-open posterior fontanel. MRI showed fused lateral ventricles with the appearance of a monoventricle which was abnormally dilated, small fourth ventricle, and no evidence of any obstructive hydrocephalus or any periventricular CSF resorption. Moreover, she had swollen lids, mucus discharge from both eyes and woody like red thick masses that replaced the normal mucosa of her eyes (Figure 3). At birth, it was misdiagnosed as congenital pseudomembranous adenoviral and treated by regular cleaning under local anesthesia every week with no improvement. Three months later, the patient was referred to an ophthalmologist and diagnosed with severe bilateral LC and treated medically with several eye drops including fluorometholone, heparin, and artificial tears, with moderate improvement, and no surgical intervention was performed.

(a, b) Bilateral swelling of the upper and lower eyelids with pseudomembranous conjunctivitis and woody like lesions in both eyes (yellow arrows).
The VP shunt surgery was postponed several times due to recurrent respiratory and urinary tract infections. Despite this, her HC continued to increase. The VP shunt was eventually inserted at the age of 8 months, but after 1 month, the patient was admitted as a case of meningitis due to CSF leak from the surgical wound, which was treated with meropenem and vancomycin. Two days postadmission, VP shunt was re-established due to malfunctioning.
The patient’s clinical features were found to be consistent with PLG I deficiency, confirmed by molecular genetic testing. She was found to have homozygous status for the common mutation like case 1, who is also from the same city, Bethlehem, Palestine: c.1752-1753insG (P.cys585valfsx15) mutation in exon 14 of PLG gene.
Parents are consanguineous; the patient has three brothers and one sister, all of whom are healthy. However, her father reported having a distant relative diagnosed with PLG I deficiency. The mother has a history of recurrent unexplained miscarriages. The treatment plan involves starting treatment with Ryplazim (generic name plasminogen, human-tvmh) intravenously every 2–4 days at a dose of 6.6 mg/kg. Unfortunately, due to limited healthcare resources in Palestine, the patient lacked access to PLG replacement therapy; so she received intravenous plasma (10 ml/kg) three times a week over the course of 3 months. Additionally, she continued using several eye drops consisting of toberamycin 0.3% four times daily, hyrdoxyethylcellulose 2% six times daily, and heparin sodium (5000 units/ml) four times daily with moderate improvement in pseudomembranous lesions and fewer hospital admissions. Therefore, conducting genetic and prenatal testing for this gene in future pregnancies will remain the mainstay of disease prevention.
Discussion
Severe PLG I deficiency is a multisystemic inherited disease with chronological variability, as symptoms can manifest either early or late in lifespan.9,10 LC is the most common presentation in the affected patients that are accounting for at least 81%. 1 It is a rare form of chronic pseudomembranous conjunctivitis, that results from the development of fibrin-rich, woody-like pseudomembranous lesions mainly on the tarsal conjunctiva. 11 Typically, it is preceded by conjunctival erythema and chronic tearing, followed by the development of red, yellow-white, or white masses. 6 The above clinical findings, ease of membrane peeling with minimal bleeding, along with histopathological examination confirms the diagnosis of LC. 11
Currently, there is no universally agreed-upon treatment plan. However, medical treatment with purified PLG replacement therapy is considered a promising approach, which has shown complete resolution of multiple cases, 12 but has limited availability in most health care centers and is cost-prohibitive in the developing countries. 13 Alternative treatment includes topical heparin, cyclosporine, corticosteroids, alpha-chymotrypsin, hyaluronidase eye drops, and topical or systemic fresh frozen plasma (FFP).14,15 Surgical excision and ocular lesions peeling in combination with intravenous FFP administration, local cyclosporine, amniotic membrane transplantation, topical heparin, or steroids can successfully treat LC. 14 It is worth mentioning that surgical excision alone is not preferred because it can trigger recurrence.14,16
Congenital hydrocephalus is uncommon in patients with PLG I deficiency and has been reported particularly in association with the more severe forms, representing 12% of the cases. 1 The clinical course is generally complicated by shunt thrombotic occlusion and subsequent decompensated hydrocephalus. However, the etiology of hydrocephalus nevertheless remains uncertain; it has been attributed, mainly within the fetal period, to a CSF block that may be resulting from extravascular fibrin deposits. Fibrin accumulates in the extracellular space because of ineffective fibrinolysis as a consequence of the PLG deficiency.7,17
The early diagnosis of the congenital hydrocephalus using CT or MRI may reduce the mortality secondary to obstructive hydrocephalus. 18 In spite of this, the prognosis is poor. The diagnosis can be supported by a family history if other affected siblings or family members are available. 6 Genetic study by undergoing whole exome sequencing is required to diagnose PLG I deficiency. 19 A selection of different genetic abnormalities has been recognized in the PLG gene in subjects with heterozygous, homozygous, and compound-heterozygous hypoplasminogenemia. 4
The main question in the management of hydrocephalus in PLG I deficiency is into which compartment the CSF should be drained into. The common distal drainage sites of a CSF shunt include the peritoneum and atrium, which are associated with a high disease complication rate due to inadequate absorptive capacity from peritoneum and pseudomembrane formation. These factors can lead to VP shunt malfunction. Therefore, hydrocephalus is one of the very few indications for the initial placement of a ventriculocholecystic shunt. 7 The latter is a good option due to its absorption capacity, allowing for the management of up to 1500 cc of fluid daily, apart from being an excellent bile duct drainage. 8 Weinzierl et al. reported two pediatric cases of PLG I deficiency treated with ventriculocholecystic shunt, which functioned well for 2 and 3 years, respectively, for each patient. 7 Additionally, Demetriades et al. described a case where a ventriculocholecystic shunt remained functional for nearly 4 years following a failed VP shunt in a 9-month-old female patient. Consequently, the authors recommend early management with ventriculocholecystic shunt as opposed to VP shunt. 20
Shapiro et al. conducted a clinical trial and administered plasminogen, human-tvmh, to 15 adult and pediatric subjects over 48 weeks. All subjects showed a 50% improvement. Moreover, a 10% increase in PLG activity level was observed after 12 weeks of treatment. 21 Based on this clinical trial, the Food and Drug Administration approved purified plasminogen, human-tvmh, marketed as Ryplazim. It is administered intravenously every 2–4 days at a dose of 6.6 mg/kg of body weight. This treatment is considered the treatment of choice for the resolution and prevention of recurrence for all lesions associated with PLG I deficiency.22,23
PLG replacement therapy is not readily available in Palestine, along many other developing countries, due to cost or supply issues, necessitating the use of alternative treatments. However, according to the literature, daily plasma transfusions (10 ml/kg) were reported to raise PLG activity to steady-state levels of 21%–22%. Biesinger et al. stated that over 4 weeks, plasma transfusion was administered, and decreased from three times per week to once per week, which resulted in a 4%–8% rise in PLG activity above baseline. 24 In case 2, our patient received three doses of intravenous plasma (10 ml/kg) over the course of 3 months and improved significantly. As a part of our approach, it will continue to be administered three times per week until PLG replacement treatment becomes available.
In a retrospective study conducted between January 2006 and January 2017 in Turkey, 17 patients (8 females and 9 males) were diagnosed with PLG deficiency. These patients received treatment with FFP, either locally or intravenously. Among the 13 patients treated using different approaches, 8 showed clinical improvement. Specifically, 11 patients received FFP intravenously and via eye drops: 6 had a good response, 3 showed no improvement, and 2 were newly treated with no documented response. Additionally, one patient received FFP only and experienced symptomatic improvement, while another patient received intravenous and bronchial FFP, with a positive response. Notably, male patients responded better and earlier to treatment than females. Overall, FFP treatment demonstrated a clinically significant 72% response rate. 25
In ultrarare diseases, registries of case reports and series have shown multiple effects on understanding the course and outcomes of various interventions, significantly impacting the prognosis and patient outcomes. There are many advantages to national, regional, and international registries for identifying the full spectrum of disorders and gaining broad insights into therapeutic interventions and patient outcomes. However, international registries is preferred, they require continuous long-term follow-up, which can be challenging due to enormous financial, technical, and human resource demands. 26
Therefore, starting with national and regional registries—especially in developing countries and the Middle East, where consanguineous marriages are more common and impact disease prevalence—can encourage health care providers to document the natural history, develop clinical guidelines, introduce new interventions, and optimize treatment regimens. 27
Conclusion
Congenital hydrocephalus followed by LC is rare and represents the severe form of PLG I deficiency, which is a genetic multisystemic condition associated with inflamed growths on the mucous membranes. PLG I deficiency carries high mortality and morbidity. Promising results are expected with the approved plasminogen, human-tvmh by the Food and Drug Administration. However, access to the newly approved drug in developing countries is challenging, often hindered by cost or supply issues, necessitating the use of alternative treatments.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics approval
Our institution does not require ethical approval for reporting individual cases or case series.
Informed consent
Written informed consent was obtained from a legally authorized representatives for anonymized patient information to be published in this article. Patient’s family consent was obtained.