
Abstract
MRKH syndrome, or Mayer-Rokitansky-Küster-Hauser syndrome, a rare congenital disease, manifests as a complete or partial aplasia of the uterus and the vagina’s upper two-thirds with normal external genitalia and functioning ovaries. Mayer-Rokitansky-Küster-Hauser syndrome can occur in isolation (type I) or in conjunction with other congenital extragenital deformities affecting the kidneys, skeleton, heart, eyes, or auditory system (type II). The diagnosis of Mayer-Rokitansky-Küster-Hauser syndrome typically relies on imaging studies, with transabdominal ultrasonography serving as the primary modality. However, magnetic resonance imaging is considered the gold standard for detailed assessment of internal genital anatomy. We present the case of an 18-year-old woman without any notable medical history who exhibited primary amenorrhea. Mayer-Rokitansky-Küster-Hauser syndrome type II was suspected on pelvic ultrasound and subsequently confirmed via magnetic resonance imaging. The patient was provided with psychological assistance and planned for vaginoplasty.
Introduction
MRKH syndrome, or Mayer-Rokitansky-Küster-Hauser syndrome, a rare congenital disease, occurs in approximately 1/4500–5000 live female births.1,2 It is characterized by a complete or partial aplasia of the uterus and the vagina’s upper two-thirds due to Müllerian duct dysgenesis. As the ovarian function remains intact, it manifests as a primary amenorrhea alongside normal secondary sex characteristics.2,3
MRKH syndrome may present as an isolated condition (type I) or in conjunction with other congenital extragenital abnormalities affecting the kidneys, skeleton, heart (atrial septal defects, aortopulmonary window, Tetralogy of Fallot and pulmonary valvular stenosis), eyes, or auditory system (type II).2,4 Ultrasound (US) is the primary imaging modality used for investigating the female pelvis, but magnetic resonance imaging (MRI) provides a highly sensitive and noninvasive means of assessing pelvic anatomy. 3
We report a case of MRKH syndrome type II featuring aplasia of the uterus and the vagina’s upper 2/3 with crossed fused renal ectopia.
Case report
MA, an 18-year-old college student, presented to the consultation for primary amenorrhea as her main complaint. The patient had no specific antecedent. There was no documented occurrence of amenorrhea in any first- or second-degree relatives. She exhibited normal development of secondary sexual characteristics with no history of cyclic pelvic pain. Physical examination showed normal external genitalia and Tanner stage 5 for the breast, pubic, and axillary hair development.
Laboratory data and hormonal evaluation (follicle-stimulating hormone (FSH), luteinizing hormone (LH), progesterone, prolactin, and testosterone) were normal. The chromosomal analysis showed a 46, XX karyotype.
A pelvic ultrasound examination (Figure 1) disclosed the agenesis of the uterus with normal follicular ovaries and two fused ectopic kidneys in the pelvic cavity. The remainder of the abdominal ultrasound was unremarkable. A pelvic MRI (Figure 2) was performed to give more precise information and identify other malformations. MRI confirmed the agenesis of the uterus and the vagina’s proximal two-third. The ovaries were normal and found in the iliac fossae. An additional finding of two fused kidney structures in the pelvis was also observed on the MRI, indicating crossed fused renal ectopia.
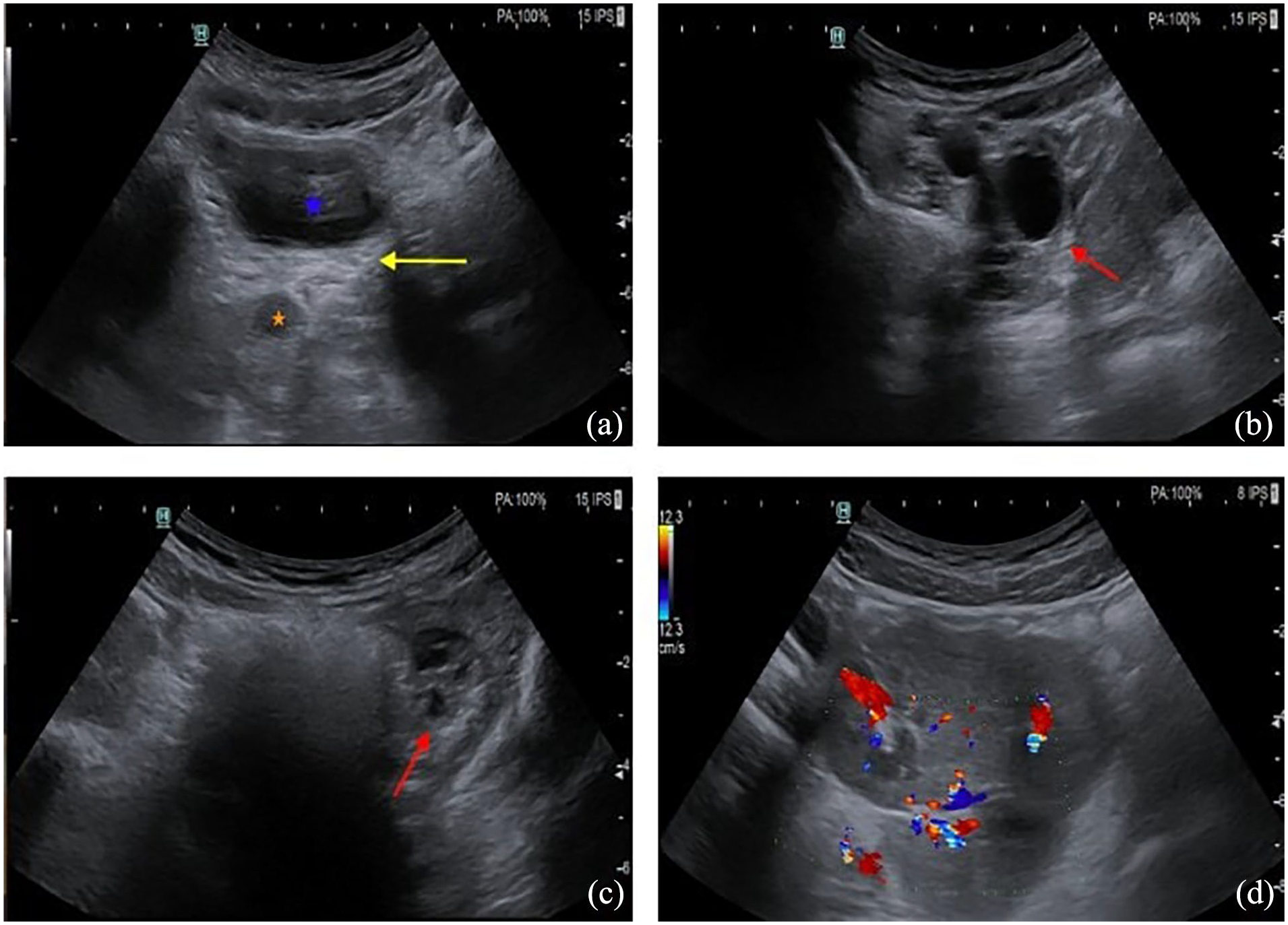
Axial Pelvic ultrasound images reveal (a) the lack of uterine tissues in the space between the bladder (blue asterisk) and the rectum (orange asterisk), (b) and (c) two normal ovaries (red arrows) in the iliac fossae and (d) fused kidneys in the pelvis.
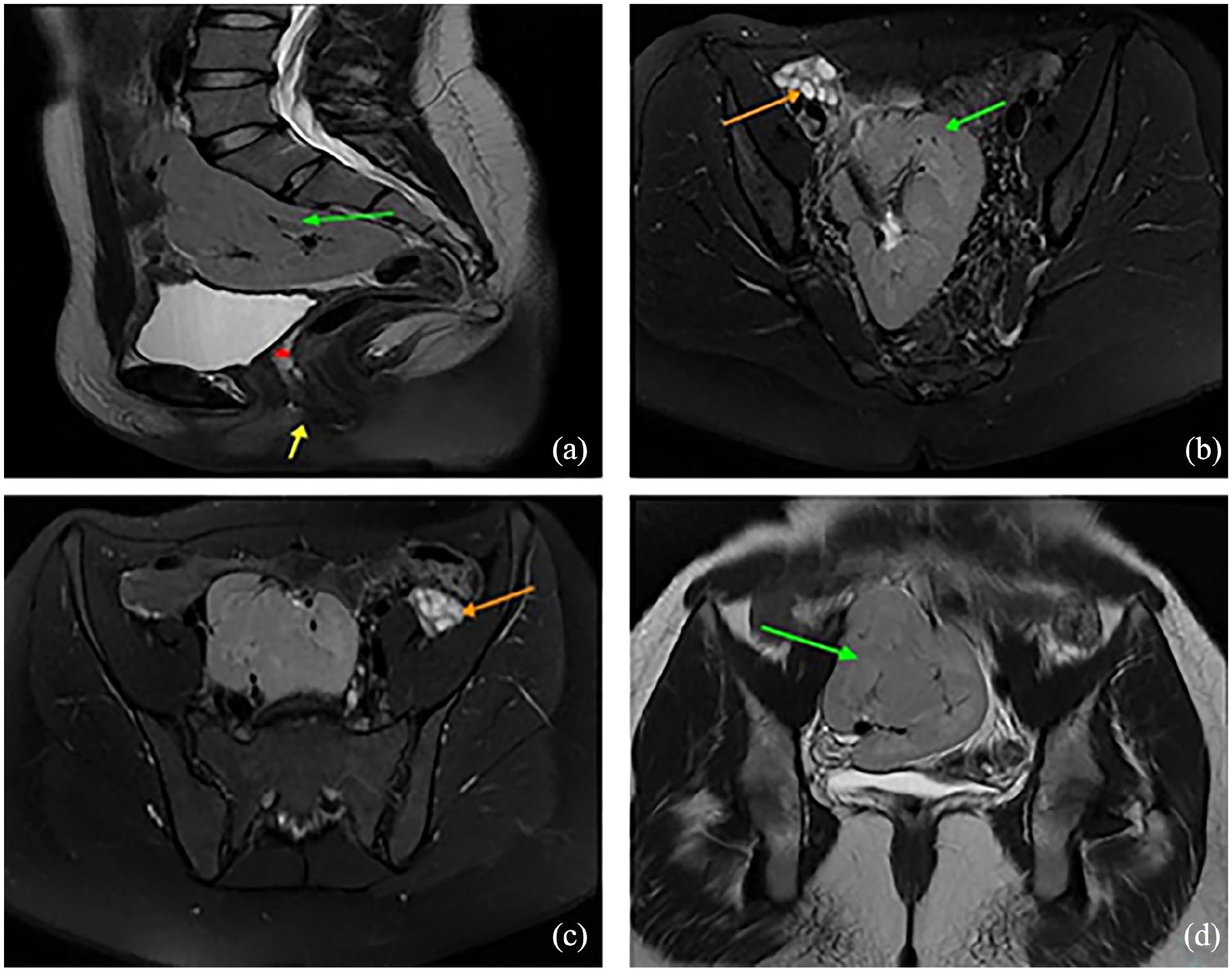
Pelvic MRI, Sagittal T2w image (a), axial T2w FS images (b) and (c), coronal T2w image (d), objectifying the lack of the uterus (red asterisk), and upper two-thirds of the vagina (yellow arrow). Each ovary (orange arrow) was in the ipsilateral iliac fossa and exhibited sub-centimetric follicles. Both kidneys (green arrow) are fused in the right iliac fossa, displaying an oval, lobulated appearance consistent with crossed-fused ectopia.
The patient’s diagnosis was determined to be MRKH syndrome type II based on these findings.
The patient was informed about the syndrome, its evolution, future management, and implications for sexual activity. In addition, she received psychological support and was scheduled for vaginoplasty as a potential option to restore sexual function.
Discussion
MRKH syndrome, known as Müllerian agenesis, was initially documented in 1829. 8 Following gonadal dysgenesis (incomplete or defective formation of the ovary or testis resulting in gonadal dysfunction), MRKH syndrome ranks as the second most common etiology of primary amenorrhea with an estimated occurrence of approximately 1/4500–5000 live female births.2,5
This syndrome is attributed to an interruption in the development of the Müllerian ducts. In typical development, between the fourth and twelfth week of gestation, a distal fusion of the Müllerian ducts forms the vagina’s upper two-thirds and the uterus, while the proximal part of the Müllerian ducts still separates, giving rise to the fallopian tubes. 5 The exact cause of MRKH syndrome is still not fully understood. The development of the Müllerian ducts, kidneys, and bone structure involves the regulation of multiple genes. It is hypothesized that irregularities in the expression of genes such as HOXA and WNT4 are significant factors leading to developmental abnormalities in the female internal genitalia.2,6
MRKH syndrome is often identified during adolescence in females exhibiting primary amenorrhea alongside the typical development of secondary sexual characteristics.7,8 Cyclic abdominal pain and dyspareunia/apareunia are observed only in uncommon cases where uterine remnants contain active endometrium. In some cases, younger children may be presented following the fortuitous detection of vaginal or uterine aplasia. 7
The physical examination shows normal external genitalia, including the vaginal dimple, the hymen, the labia minora, the labia majora, and the clitoris. In addition, the examination indicates Tanner stage 5 for breast, pubic, and axillary hair development.5,6
Patients with MRKH syndrome typically possess a 46, XX karyotype. Their hormone profile, including levels of FSH and LH, remains normal, indicating normal ovaries in both structure and function, thereby eliminating hypothalamic-pituitary axis abnormalities. Normal levels of dehydroepiandrosterone sulfate, testosterone, aldosterone, estradiol, and cortisol indicate proper functioning of the adrenal glands.5,9
Due to its accessibility, transabdominal ultrasonography is commonly chosen for assessing Müllerian duct abnormalities, but its effectiveness can vary depending on the operator’s skill. However, MRI is considered the gold standard for providing a detailed assessment of internal genital anatomy.3,9 The ultrasound shows the absence of uterine tissues in the space between the bladder and the rectum. 6 MRI facilitates precise assessment of uterine aplasia and provides detailed information about the ovaries’ existence, structure, and position.6,9 The assessment also involves identifying the presence or absence of remnants of the Müllerian duct, manifesting as rudimentary uterine buds. These structures are commonly located antero-inferior to their respective ipsilateral ovaries. On T1-weighted imaging, they appear as dark, solid ovoid structures; on T2-weighted imaging, they may show a cavitated appearance with a target-sign appearance. 9 MRI may uncover concurrent renal and skeletal abnormalities. 6
Laparoscopy is necessary as a final option if imaging modalities have not provided adequate information, especially in cases involving a rudimentary uterus or for the treatment of rudimentary uterine buds.5,9,10
The range of potential diagnoses encompasses obstructive anomalies, including low transverse vaginal septum, imperforate hymen, distal vaginal atresia, and cervical atresia, alongside Turner syndrome, CYP17A1 deficiency, and androgen insensitivity syndrome.4,9,11 Clinical examination, karyotype analysis, hormone profiling, and imaging studies assist in distinguishing MRKH syndrome from other conditions. Patients with obstructive anomalies typically present with cyclic pelvic pain. On digital rectal examination, they may exhibit a normal or bulging cervix and uterus, as confirmed by pelvic imaging showing hematocolpos or hematometra. 11 Androgen insensitivity syndrome, also referred to as feminization syndrome, is differentiated from Müllerian dysgenesis by several factors, including a 46, XY karyotype, elevated male testosterone levels, absence of the uterus and female adnexa, and identification of abdominal or inguinal testes on MRI.6,11 In cases where Turner syndrome is suspected due to short stature and delayed puberty, confirmation through karyotype analysis (45, XO) and elevated FSH levels is crucial.9,11 Similarly, in instances of delayed puberty in patients presenting with external female genitalia and hypertension, diagnosis of CYP17A1 deficiency is made through testing of the CYP17A1 gene and assessment of impaired adrenal hormone levels, characterized by elevated serum corticosterone and deoxycorticosterone levels, and low cortisol, estrogen, and androgen levels. 11
The management of MRKH syndrome requires a comprehensive strategy encompassing psychological and sexual care to improve psychological well-being and reduce the anxiety and mental stress the patient suffers after being diagnosed, along with the creation of a neovagina using nonsurgical techniques such as the method of Franck’s dilator and surgical procedures like the Abbe-McIndoe operation, the Vecchietti operation, and sigmoidal colpoplasty to improve sexual function.4,6,10
Conclusion
MRKH syndrome is a rare congenital disorder marked by varying degrees of uterovaginal agenesis and typically functional ovaries. Accurate diagnosis is vital for effective management. In this case, the patient exhibits MRKH syndrome type II, initially suspected on transabdominal ultrasound and confirmed via MRI. This highlights the critical role of radiological imaging in diagnosis, classification, and surgical planning. With its exceptional soft tissue resolution, MRI emerges as the gold standard for confirming the diagnosis, precisely evaluating pelvic anatomy, uterovaginal aplasia, ovarian location, and associated malformations.
Footnotes
Acknowledgements
I would like to express my gratitude to my professors and all the colleagues who participated in the completion of this work.
Author contributions
K.S. conception of the work, design of the work, and acquisition of data; S.S. and K.M. acquisition of data; F.C. contributed to acquisition, analysis, or interpretation; K.I., N.B., and I.N. revising the work critically for important intellectual content.
Ethical approval
Our institution does not require ethical approval for reporting individual cases or case series.
Informed consent
Written informed consent was obtained from the patient(s) for their anonymized information to be published in this article.
Guarantor of submission
The corresponding author is the guarantor of submission.