
Abstract
Chronic recurrent multifocal osteomyelitis is a rare auto-inflammatory disease in children, with only a few reports of its association with other inflammatory diseases, such as systemic juvenile idiopathic arthritis. A 15-year-old boy was admitted due to fever, skin rash, arthritis, and high inflammatory factors and was finally diagnosed with systemic juvenile idiopathic arthritis. After 6 months of recovery from the disease, the patient was referred due to local pain and swelling in the arms and left thigh. In radiography, bone lesions were seen in the shoulders, left humerus, and left femoral diaphysis. A whole-body bone scan showed increased absorption in these areas, which suggested a tumor or osteomyelitis. A biopsy of the bone lesion of the left humerus confirmed sterile osteomyelitis. Although the co-incidence of chronic recurrent multifocal osteomyelitis with systemic juvenile idiopathic arthritis is rare, it should be considered in differential diagnosis.
Introduction
Chronic recurrent multifocal osteomyelitis (CRMO) is a rare condition occurring in 1–2 per 1,000,000 cases. This condition mainly occurs in the early ages of life; however, its presence in adulthood is not uncommon. 1 CRMO combines various presentations, including synovitis, acne, pustulosis, hyperostosis, and osteitis (SAPHO), which constitute a unique disease spectrum of chronic nonbacterial osteomyelitis (CNO). Therefore, CRMO is characterized by its related symptoms, bone pain with or without fever, in conjunction with radiological studies indicating the mixture of osteolytic or sclerotic lesions. CRMO has an unpredictable periodic manner alternating between spontaneous remissions and exacerbations. 2
The concurrent incidence of CRMO with other autoimmune or inflammatory conditions can be of great clinical interest, which has led to the categorization of this disorder in the group of systemic juvenile idiopathic arthritis (sJIA) or juvenile spondyloarthropathy. However, evidence in this regard is limited and inconclusive. 3 The current study aims to present a case with sJIA who developed CRMO in a short period after juvenile idiopathic arthritis-related symptoms were controlled.
Case presentation
A 15-year-old boy was admitted to our hospital, Imam Hossein Children’s Hospital, Isfahan University of Medical Sciences with the chief complaints of fever, arthritis, and cutaneous rash. The condition was initiated 3 weeks ago via hectic fevers, mainly in the evenings and at night. The patient turned ill during the fevers, and salmon-colored rashes appeared on his proximal extremities, trunk, and abdomen. By the fever control, the patient was asymptomatic, the rashes disappeared, and the patient’s general condition improved. Besides, during this period, articular inflammation was noticed in both knees, ankles, and the left elbow.
Despite recurrent referrals to outpatient clinics and receiving antipyretic agents, his symptoms did not recover. Accordingly, he was admitted to the rheumatology ward. In the family history, there was no history of autoimmune diseases, and his parents were not related.
His fever was checked every 6 h, any rash was recorded if it appeared, and the joints were examined routinely to assess the presence of any inflammation.
Considering the spiking evening and nocturnal fevers, rash, and multiarticular arthritis for 2 weeks, the diagnosis of sJIA was considered, and paraclinical information was requested, which demonstrated leukocytosis white blood cells: 25,000/mm3), thrombocytosis (platelet: 730,000/mm3), neutrophilia (neutrophil ratio: 90%), estimate erythrocyte sedimentation (ESR: 110 mm/h), and C-reactive protein (58 mg/dl). Blood culture, urine culture, brucellosis serology, COVID-19 polymerase chain reaction, cytomegalovirus antibodies, Epstein barre virus antibodies, and Parvovirus B19 serology were all negative. Rheumatologic tests including angiotensin-converting enzyme, antinuclear antibody, and antiphospholipid antibodies, and human leukocyte antigen-B5 (HLA-B5), HLA-B51, and HLA-B27 were negative. Complement tests and echocardiography were normal. Bone marrow aspiration revealed normal entities with no evidence of malignancy. Based on the clinical and paraclinical manifestations and ruling out the other potential diagnoses, according to the International League of Associations for Rheumatology (ILAR) criteria, sJIA diagnosis was confirmed.
Accordingly, the medication with methylprednisolone corticosteroid pulse therapy (30 mg/kg for 3 days), naproxen (20 mg/kg/day), and methotrexate (15 mg/m2/day) was initiated. Within 5 days, the symptoms disappeared, inflammation-associated laboratory parameters improved, and the patient was discharged with the therapeutic order of prednisolone (1 mg/kg/day), methotrexate (15 mg/m2/day), and naproxen (20 mg/kg/day).
He was followed for a month, during which time his general condition was acceptable, and no evidence of articular inflammation, cutaneous rash, or organomegaly was noticed. Therefore, the corticosteroid treatment was tapered, and the other medications were continued.
In the next visit, within 3 months, the daily prednisolone was reduced to 5 mg/day, and methotrexate turned from the injecting route to oral tablets, while naproxen was continued with the same pattern.
Within the next 6 months, the patient was referred with significant pain complaints and swelling in the shoulders, left arm, and thigh. The physical examination was regular, and there was no evidence of arthritis or enthesitis. Plain radiography was taken from the mentioned bones, which contained bone lesions suggestive of tumoral changes or osteomyelitis (Figure 1).
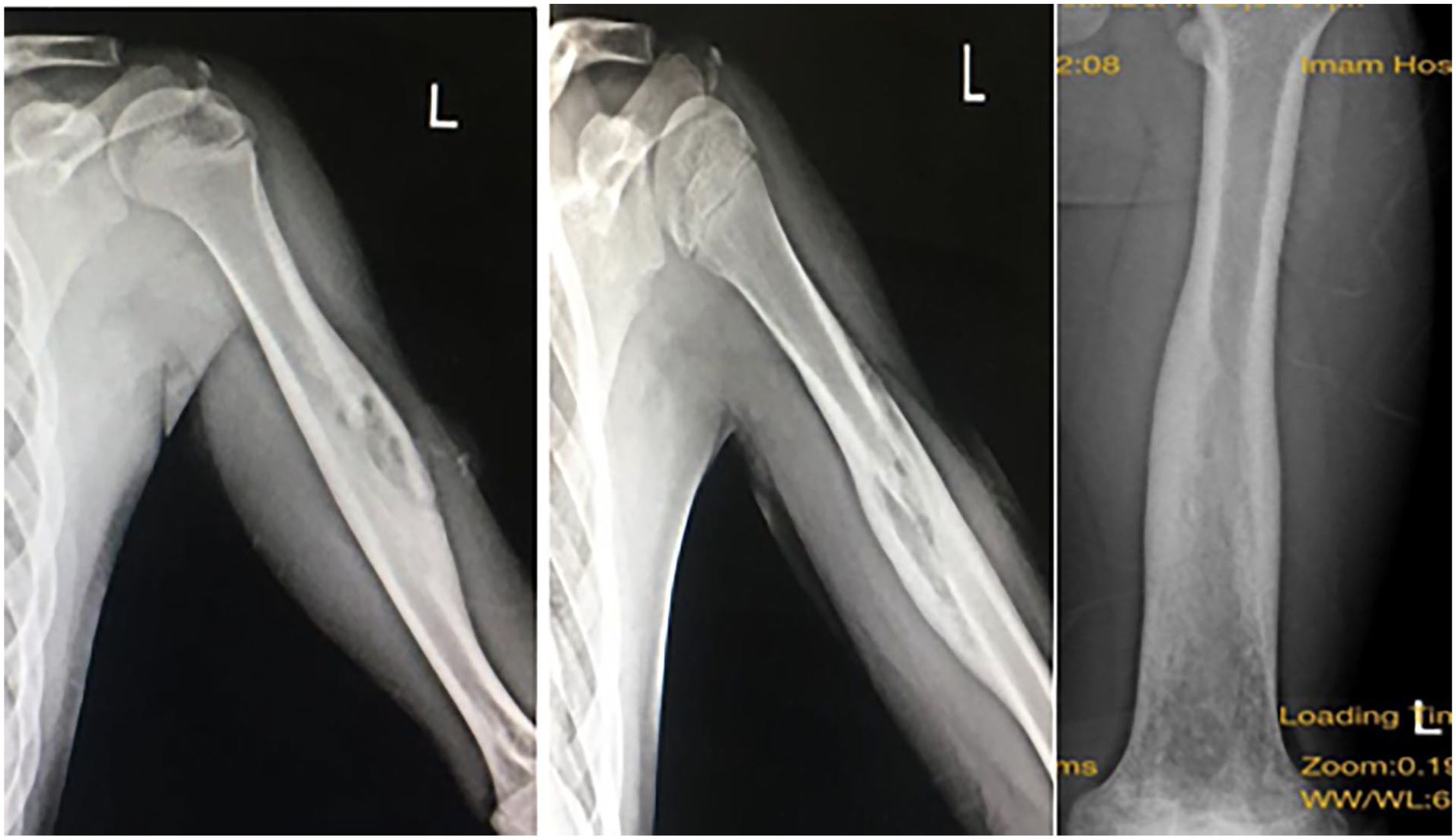
Plain radiography: bone lesions in the left humerus, and left femoral diaphysis.
A whole-body bone scan represented increased absorption in the shoulders, left humerus, and left femoral diaphysis, suggesting inflammatory or malignant lesions (Figure 2).

A whole-body bone scan: increased absorption in the shoulders, left humerus, and left femoral diaphysis.
Inflammatory laboratory results were in normal ranges. Therefore, biopsies were taken from the left humerus diaphysis, which showed mixed inflammatory infiltrate, including histocytes, plasma cells, lymphocytes, and a few neutrophils, without any documentation of malignancy or infection. The bone culture was also negative and confirmed sterile osteomyelitis. The diagnosis of CRMO was confirmed by obtaining all documents and findings.
Discussion
sJIA is one of the categories of JIA classified by the (ILAR). Daily spikes of fever, arthritis, salmon-colored rashes, generalized lymphadenopathy, and organomegaly are the significant manifestations of sJIA, 4 all noted in our patient.
The pathogenesis of sJIA is compatible with an auto-inflammatory syndrome, considering the facts about innate immunity dysregulation plus the absence of autoantibodies. 5 Blocking biologics of IL-1 β, IL-6, or TNF-α-inhibiting agents shows promise in controlling sJIA symptoms. 6
CRMO or CNO was first described by Giedon and colleagues in 1972 when an unusual subacute and chronic symmetrical osteomyelitis was notified in a patient with multifocal lesions. 7 The investigations regarding the diagnosis of these disorders were initiated, and it was concluded that CRMO could be marked when the other probable differential diagnoses associated with bone inflammation, malignancies, and bone-involving bacterial infections have been ruled out. Therefore, radiological, laboratory, and bone marrow biopsy investigations are required in the primary steps. 8 Accordingly, we performed all the mentioned approaches that represented no manifestation of inflammation, infection, or tumoral change.
The authors reporting this rare disorder have abundantly noted CRMO’s association with other inflammatory diseases such as inflammatory bowel disease (IBD), psoriasis, and palmoplantar pustulosis. 8 Bone disease in CRMO is an inflammasome-independent, IL-1-mediated disease. Infantile-onset multifocal osteitis associated with generalized pustulosis is due to dysregulation of the IL-1 pathway. This fact confirms that SAPHO, CRMO, and related disorders are autoinflammatory. CRMO is accompanied by some non-rheumatic medical conditions, the most common of which include allergic rhinitis, attention-deficit hyperactivity disorder, and atopic dermatitis. 12
As far as we know, this report is only the second one published about the occurrence of both CRMO and sJIA. The first report was by Arakawa et al., and their patient also had symptoms of diarrhea and fever, which led to suspicion of IBD. It was found that the patient had CRMO and IBD at the same time. Their further assessments revealed no manifestation of IBD. 9 However, Tsitsami et al. 10 represented another case with CRMO, which had concurrent manifestations of JIA but not sJIA. However, JIA cannot be exclusively excluded from the differential diagnoses of CRMO, as arthritis, morning stiffness, and asymmetrical joint involvement have been reported in a study by Beck et al., 11 who investigated 37 patients with CRMO. However, one of the significant characteristics of CRMO that differentiates this disorder from JIA-group syndromes is lacking inflammation in the joints. 12 CRMO may affect any bone of the body, but the metaphyseal regions of the long bones, clavicle, and vertebral bodies are the most commonly affected bones. Asymptomatic lesions can be screened for by a technetium bone scan. MRI is functional for showing the activity and extent of the bone lesions and is superior to bone scans for diagnosis. Whole-body MRI with short tau inversion recovery images offers an alternative to bone scan. However, we did not have the equipment and facilities to perform whole-body MRIs for this patient.
NSAIDs are the first-line therapeutic approach of CRMO; however, our patient was under naproxen therapy plus other immune regulatory medications when his symptoms initiated. Recently, different agents have been utilized to control CRMO symptoms but none has been systematically investigated or approved by drug and food organizations. 9 Treatment including disease-modifying antirheumatic drugs, colchicine, intravenous immunoglobulins, corticosteroids, and bisphospohnates. Desirable outcomes have accompanied corticosteroids; however, they are suggested to be applied in limited doses. Most of the attention has recently been turned toward biological antibodies, with TNF-alpha inhibitors at the top of the list. An increasing body of evidence suggests biological agents in combination with bisphosphonates for CRMO. 8
The auto-inflammatory nature of both diseases might overlap the symptoms, making it difficult to attribute the symptoms or response to the treatment of each disorder, sJIA versus CRMO. Most case reports of the association between these two autoinflammatory diseases have been in girls and infancy or childhood and less than 13 years1–3,13; however, our patient was a 15-year-old boy. However, our study had limitations such as the lack of genetic examination and whole-body MRI.
The important point is to always consider hematogenous multifocal osteomyelitis in the differential diagnosis of CRMO. Hematogenous multifocal osteomyelitis usually occurs in children with immunodeficiency or sepsis, and if not treated quickly, it leads to morbidity and mortality. Unlike CRMO, this disease is acute and associated with high fever, toxic general condition, hemodynamic imbalance, and loss of consciousness.14,15
Conclusion
Although rare, the possibility of CRMO co-occurring with sJIA should not be overlooked or underestimated. Although rare, the association of two or more auto-inflammatory diseases in children requires attention.
Footnotes
Acknowledgements
The authors are thankful to the patient who agreed to publish her case report.
Author contributions
M.J. conception of the work and final edit, S.A. has drafted the work. All authors read and approved the final manuscript.
Availability of data and material
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics approval
Our institution does not require ethical approval for reporting individual cases or case series.
Informed consent
Written informed consent was obtained from a legally authorized representative(s) for anonymized patient information to be published in this article.