
Abstract
Pyogenic arthritis, pyoderma gangrenosum, and acne syndrome is a rare disease, linked to an auto-inflammatory pathway. We report a 7-year-old boy with recurrent suppurative knee arthritis without signs of suppurative skin infection or ulcer; his younger brother had the same symptom. Genetic testing indicated the presence of proline-serine-threonine phosphatase interacting protein 1 gene mutation in both boys. Our patient’s grandfather had a history of recurrent pyoderma, and his father though a genetic carrier had no symptoms. Interestingly, our patient displayed markedly high levels of interleukin-6, while interleukin-1 and other cytokines were not elevated. These lab findings led to the treatment of pyogenic arthritis, pyoderma gangrenosum, and acne syndrome with tocilizumab. Previously reported cases of similar phenotypes of the syndrome have not presented in this fashion, nor have there been reported cases of pyogenic arthritis, pyoderma gangrenosum, and acne syndrome with a positive family history and an elevation in interleukin-6. The mutation site of proline-serine-threonine phosphatase interacting protein 1 in this incomplete pyogenic arthritis, pyoderma gangrenosum, and acne syndrome has not been reported before. It is possible that there are other pathogenic ways to trigger these auto-inflammatory disorders. Tocilizumab, which specifically targets interleukin-6, was effective in this case.
Background
Pyogenic arthritis, pyoderma gangrenosum, and acne syndrome (PAPA syndrome) is an auto-inflammatory disease associated with the proline-serine-threonine phosphatase interacting protein 1 (PSTPIP1) gene mutation. This gene encodes PSTPIP1, which interacts with pyrin. 1 This mutation leads to an increased affinity for pyrin, leading to up-regulation of caspase 1 and activation of interleukin-1 (IL-1), which produces a neutrophil-mediated response. 2 Thus, a whole-body abnormal immune reaction typically occurs. The clinical features of PAPA syndrome include fever, acne, pyoderma gangrenosum, joint swelling, and pain. While this syndrome is rare in China, the child reported in this article lives in Xinjiang, China, and has an incomplete presentation and positive family history.
Case report
A 7-year-old boy of Han ethnicity, weighing 23.5 kg, was admitted to hospital with recurrent episodes of joint swelling and severe pain for 1 year. He had two surgeries in the prior year for suppurative knee arthritis. There was no obvious trauma before the onset of each episode, and no pyoderma, acne, suppurative hidradenitis, systemic infection, or fever during the course of the disease. Physical examination showed that the right knee joint of the child was more swollen than the left, with higher skin temperature and tenderness, but no obvious sense of fluctuation upon palpation. His right knee movement was restricted with flexion less than 40° and stretching up to 0°. Internal and external rotation of the right knee joint was also slightly limited and the floating patella test was negative. No abnormalities were found in the other limb joints.
At the time of admission, the total number of white blood cells (11.08 × 109/L) in the peripheral blood was high, mainly comprising neutrophils (7.89 × 109/L, 71.2%). The erythrocyte sedimentation rate (ESR) was elevated (73 mm/h). Liver and kidney function showed no obvious abnormality. C-reactive protein (CRP, 123 mg/L), procalcitonin (PCT, 0.10 ng/mL), and interleukin-6 (IL-6, 73.25 pg/mL) were all increased. The levels of interleukin-1β (IL-1β, <5.0 pg/mL), interleukin-8 (IL-8, 15.6 pg/mL), and interleukin-10 (IL-10, <5.0 pg/mL) were normal. Rheumatoid factor (RF) antibody levels (IgG 26.08 AU/mL, IgA 4.12 AU/mL, and IgM 2.41 IU/mL) were within the normal range, too. Autoimmune antibodies such as anti-keratin antibodies (AKA), antinuclear antibodies (ANA), anti-double-stranded DNA (anti-dsDNA) antibodies, anti-Smith antibodies, and anti-neutrophil cytoplasmic antibodies (ANCA, including p-ANCA, c-ANCA, myeloperoxidase, and proteinase-3) were all negative, as was human leucocyte antigen (HLA-B27). Articular cavity effusion was thick and turbid with a high number of nucleated cells (3+/HPF) and pus cells (1+/HPF), 98% of which were multi-nucleated cells. No bacterial growth was observed in either culture of articular cavity effusion (microbiology cultures were saved for 28 days). The patient’s blood coagulation function and viral screening (human immunodeficiency virus (HIV), syphilis, and hepatitis) were normal.
The boy underwent arthroscopic knee debridement and peri-knee abscess resection. He was treated with vancomycin and rifampicin after each operation, but his arthritis continued to intermittently attack. Upon further inquiry, we found that the patient’s 5-year-old brother had the same history of arthritis in his right elbow and left ankle. The boy’s grandfather also had a history of recurrent pyoderma. We recommended that the whole family undergo genetic testing, which revealed that the patient, his brother, and his father were all heterozygous for the PSTPIP1 gene mutation, which was found in the exon region (c.36+68 G>A, c.137+47 G>C, and c.562+114C>G het). These three mutated sites have not been reported in the previous literature, among which the first and second mutated sites were highly expressed in the sequencing of 1000 normal subjects, while the third mutated site was not reported in the normal genome database, so it may be a possible deleterious mutated site. Although the father was a carrier, he never showed signs of the syndrome. The grandfather declined genetic testing. After the diagnosis of PAPA was made, the boy was treated with naproxen (0.2 g per time, four times a day), high-dose prednisone (1–2 mg/kg/day), and methotrexate (10 mg/m2/week). However, due to poor efficacy of the medication combination and elevation of IL-6, we switched to intravenous infusion of tocilizumab once every 4 weeks at a dose of 8 mg/kg. The boy had received four treatments with tocilizumab and did not develop suppurative arthritis for the duration of treatment. The CRP (27.40 mg/L), PCT (0.04 ng/mL), and IL-6 (23.88 pg/mL) levels were restored or nearly normal.
Discussion
PAPA syndrome, first reported in 2012, is one of the auto-inflammatory diseases. 3 Given the association of PSTPIP1 mutations found in these syndromes with similar phenotypes (PAPA; pyoderma gangrenosum, acne, and suppurative hidradenitis (PASH); axial spondyloarthritis with the triad of PASH (PASS); pyoderma gangrenosum, acne, and ulcerative colitis (PAC)), they may belong to the same pathogenic spectrum. 4 From a pathophysiological standpoint, all of these syndromes share a common mechanism consisting of over-activation of the innate immune system, leading to increased production of the IL-1 family and “sterile” neutrophil-rich cutaneous inflammation. 5 IL-1 or tumor necrosis factor (TNF) receptor antagonists are typically selected as treatment for this group of related conditions.5–7
However, the child we report varied from previously reported cases of this rare syndrome. He had only recurrent episodes of suppurative arthritis, but no fever, acne, or pyoderma gangrenosum; his younger brother presented in the same way. Interestingly, the patient displayed a markedly high level of IL-6, while his IL-1 and other cytokines were not elevated. The two boys’ clinical manifestations were also different from those of their grandfather, who only had recurrent pyoderma, while their father, who also carried the gene mutation, exhibited no symptoms. Previous literature has also mentioned the existence of incomplete penetrance and expressivity. 8 These differences may all stem from the heterozygosity of the mutated PSTPIP1 gene, and the mutated site had not been previously reported, which affects the proteins of the inflammasome complex (the molecular platform responsible for triggering auto-inflammation) or the proteins that regulate inflammasome function. Tocilizumab, a biologic agent specifically targeting IL-6, is currently the most effective treatment.
Although this boy’s clinical presentation is incomplete, possibly due to young age of both brothers, we still tend to diagnose him as having an auto-inflammatory disease such as PAPA syndrome rather than juvenile idiopathic arthritis (JIA) for several reasons. (1) The child’s clinical manifestation was recurrent sterile pyogenic arthritis, which rarely appeared in JIA, but was common in PAPA. (2) Autoimmune antibodies such as AKA, ANA, anti-dsDNA antibodies, anti-Smith antibodies, and ANCA were all negative, as were RF and HLA-B27, so other autoimmune rheumatic diseases, including JIA, were not supported. (3) The similarity of the two brothers’ symptoms, their grandfather’s recurrent pyoderma, and the discovery of PSTPIP1 gene mutation all support the diagnosis of PAPA syndrome or another auto-inflammatory disease similar to PAPA. Maybe we can call it incomplete PAPA syndrome with elevated IL-6. PSTPIP1-associated myeloid-related proteinemia inflammatory syndrome (PAMI), another kind of auto-inflammatory disease, could also be eliminated because of the absence of neutropenia, anemia, thrombocytopenia, and liver or spleen enlargement.
In addition to PAPA syndrome, other major single-gene auto-inflammatory diseases are listed in Table 1. Because of low incidence and varying clinical manifestations, the understanding of newly discovered auto-inflammatory diseases is limited, making diagnosis difficult. There are, however, some rules and procedures to diagnose such diseases. Although the clinical manifestations of these diseases are diverse and overlapping, they do each have their own characteristics (Figure 1). In addition, an auto-inflammatory disease that first presents with chronic arthritis can be screened for a specific diagnosis or syndrome according to the following flowchart (Figure 2).
The classification of major single-gene auto-inflammatory diseases.

PA-PASH: pyoderma gangrenosum, acne, pyogenic arthritis, and suppurative hidradenitis syndrome; TRAPS: tumor necrosis factor receptor-associated periodic fever syndrome; CAPS: cryopyrin-associated periodic syndrome; Blau syndrome: familial juvenile systemic granuloma; FMF: familial Mediterranean fever; MKD: mevalonate kinase deficiency; DIRA: interleukin-1 receptor antagonist deficiency; CANDLE syndrome: chronic atypical neutrophilic skin disease with lipid dystrophy and fever; TNF: tumor necrosis factor; PSTPIP1: proline-serine-threonine phosphatase interacting protein 1; TNFRSF1A: tumor necrosis factor receptor super family 1A; NLRP3: NACHT, LRR, and PYD domains-containing protein 3; NOD2: nucleotide-binding oligomerization domain-containing protein 2; MEFV: Mediterranean fever; MVK: mevalonate kinase; IL-1RN: interleukin-1 receptor antagonist; PSMβ8: proteasome β8; AD: autosomal dominant; AR: autosomal recessive; ROS: reactive oxygen species; MARK: mitogen-activated protein kinase; NF-κB: nuclear factor-kappa B; ASC: apoptosis-associated speck-like protein containing CARD (cysteine proteases activate and recruit domains); FACS: familial cold auto-inflammatory syndrome; HIDS: hyperimmunoglobulinemia D with periodic fever syndrome; EOS: early-onset sarcoidosis.

Clinical differentiation of common auto-inflammatory diseases.
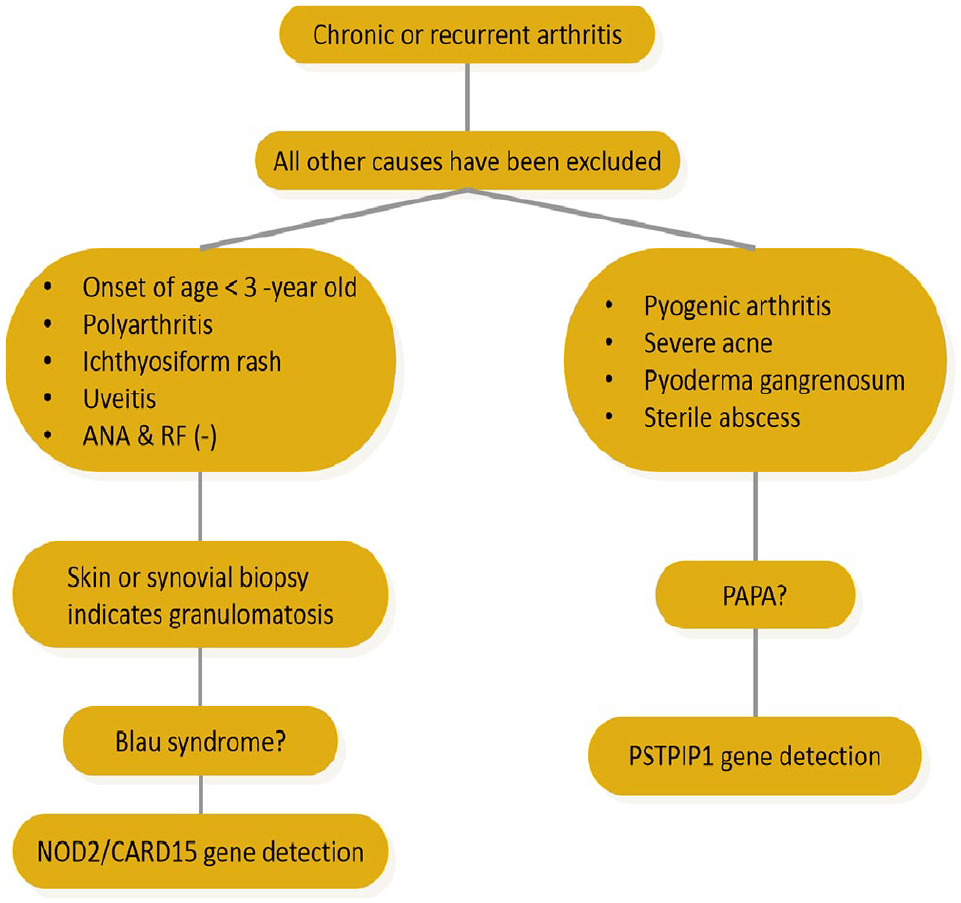
Diagnosis process of PAPA.
Conclusion
Previously reported cases of similar phenotypes of the syndrome have not presented in this fashion, nor have there been reported cases of PAPA syndrome with a positive family history and an elevation in IL-6. The mutation site of PSTPIP1 in this incomplete PAPA syndrome has not been reported before. The reason of elevation in IL-6 is currently unclear, but it is possible that there are other pathogenic ways to trigger these auto-inflammatory disorders. Tocilizumab, a biologic agent specifically targeting IL-6, was effective in the treatment of our patient.
Footnotes
Acknowledgements
Declaration of conflict of interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Informed consent
Written informed consent was obtained from the minor patient’s a legally authorized representative for anonymized patient information to be published in this article.