
Abstract
Congenital mesoblastic nephroma is considered a tumour with favourable clinical behaviour with only few reported cases of metastases. We report an infant who underwent complete resection and later developed pulmonary metastasis. Ten-month-old baby girl initially presented at 3 weeks of age with macroscopic haematuria, hypertension and a lumbar mass. Contrast-enhanced computed tomography revealed a tumour arising from the left kidney without local invasion or metastasis. She underwent left nephrectomy. Immunohistochemistry confirmed a cellular type of congenital mesoblastic nephroma. At 10 months, she presented with difficulty in breathing. Contrast-enhanced computed tomography revealed an opacity in the right hemi-thorax. Histology of lung mass was suggestive of deposits from the previously excised mesoblastic nephroma. She developed a right-sided haemothorax and succumbed. This case report highlights the fact that even though congenital mesoblastic nephromas are considered tumours with favourable clinical behaviour, they can present later with distant metastasis. Therefore, clinicians need to be aware of this rare malignant potential and adhere to meticulous follow-up protocols.
Introduction
Congenital mesoblastic nephroma (CMN) is a mesenchymal tumour in children that has been distinguished as a unique entity from Wilms tumour about half a century ago. 1
It is the commonest solid renal tumour identified in the neonatal period and represents 3%–10% of all renal tumours in children with a peak incidence during the first 3 months of life. 2 More than 90% of CMN are diagnosed within the first year. 3 Three histological subtypes of CMN have been described: classic, cellular (aggressive type) and mixed type. 4
Although generally considered a tumour with favourable clinical behaviour, rare malignant variants of CMN have been reported with metastasis to the lungs, liver, heart and brain. 5 We report a Sri Lankan infant who underwent complete resection of a mesoblastic nephroma during the neonatal period and presented during late infancy with metastatic deposits in the lungs.
Case presentation
This 10-month-old baby girl had an uncomplicated antenatal period and was born by an elective lower segment caesarean section at term. Routine antenatal ultrasound scans did not reveal any abnormalities, including a renal mass, polyhydramnios or hydrops. The immediate postnatal period was uncomplicated as well, and the baby had been discharged on day 3 of life without evidence of abdominal distension or respiratory distress.
At 1 week of age, parents had noted abdominal distension, and by the second week of life, two episodes of macroscopic haematuria prompted them to seek medical attention. On evaluation, she was found to be pale and hypertensive with blood pressure above the 99th percentile. 6 Abdominal examination had revealed a mass in the left lumbar region.
Ultrasound scan of the abdomen demonstrated a retroperitoneal mass antero-medial to the left kidney. Contrast-enhanced computed tomography (CT) revealed a renal mass arising from the upper pole of the left kidney without focal invasion or distal metastasis suggestive of stage I CMN (Figure 1). At 1 month of age, she underwent left-sided nephrectomy with curative intent, resulting in complete resection of the renal tumour and mesenteric lymph node excision.
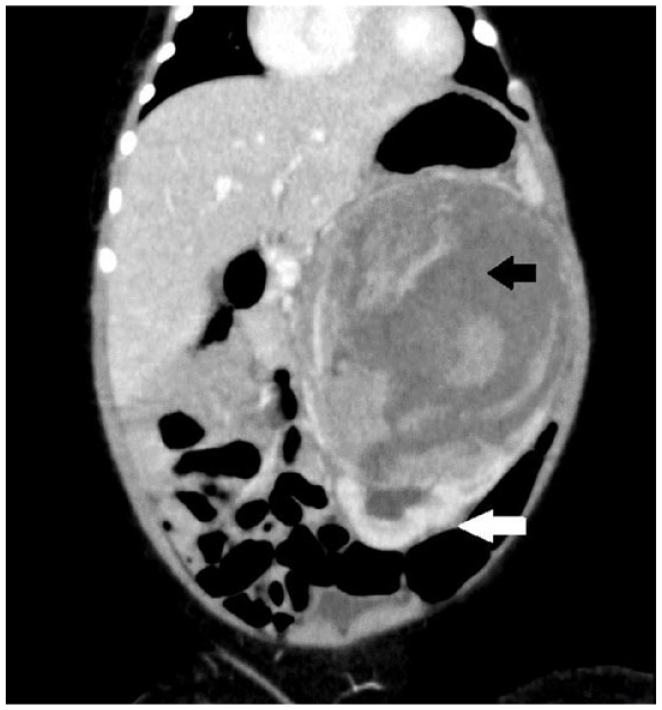
Contrast-enhanced CT of the abdomen showing renal tumour arising from the superior pole of the left kidney. (White arrow – lower pole of the kidney, black arrow – tumour with a necrotic centre.)
Histological examination revealed sections of normal kidney infiltrated by spindle cells arranged in fascicles, which showed scanty cytoplasm, moderately pleomorphic nuclei with numerous mitotic figures. Interspersed vessels appeared erratic and thin-walled with a hemangiopericytoma-like vascular pattern. Glomeruli were entrapped among the tumour cells with no evidence of necrosis (Figure 2). Resected mesenteric lymph nodes and the specimen margins were free of tumour cells (R0). Differential diagnoses from a histological perspective included clear cell sarcoma of the kidney and CMN.

Histological specimen – H&E staining of the nephrectomy specimen showing cellular proliferation and spindle cells.
Immunohistochemical staining (Figure 3) showed positivity for CD99, Vimentin and a Ki-67 index of 45% with negative staining for Desmin, Caldesmon, SMA (Smooth Muscle Actin), MyoD1, Cyclin D1, CD34, WT1 (Wilms Tumour 1), NSE (Neuron-Specific Enolase), CK (Cytokeratin) and NF (Neurofilament). Molecular genetic studies with ArcherTM FusionPlex Pan-Solid Tumour Next-Generation Sequencing Assay revealed Epidermal Growth Factor Receptor (EGFR) internal tandem duplication, confirming a cellular type CMN with BCOR (BCL6 corepressor) and pan-TRK (tyrosine receptor kinase) negativity.

Immunohistochemistry of the renal biopsy showing strong diffuse CD 99 positivity.
The infant was followed up regularly, looking for development of abdominal distension, haematuria or respiratory distress. Abdominal ultrasound scans performed every 6 weeks were persistently normal. As there were no signs of respiratory system involvement, routine serial lung imaging tests were not carried out during this follow-up period. Her growth and development followed age-appropriate limits, and she remained asymptomatic and normotensive.
At 10 months of age, the baby was noted to have gradual onset progressive difficulty in breathing. She developed vomiting after feeds and poor sucking with resting dyspnoea. Her weight began to falter, and she deteriorated progressively. On examination, she was tachypnoeic, with reduced chest expansion on the right side and dull percussion note, reduced vocal resonance and diminished air entry. Chest X-ray demonstrated an opacity in the right hemi-thorax with tracheal deviation to left side (Figure 4). Contrast-enhanced CT chest showed a large heterogeneous dense soft tissue mass with contrast enhancement and necrotic regions in right hemi-thorax measuring 8.6 cm × 6 cm × 7 cm with mediastinal shift to the left side. Right lower lobe bronchus was compressed by the mass with collapse consolidation of the right lower lobe (Figure 5).

Chest X-ray showing opacity in the right hemi-thorax with irregular margins, occupying the middle and lower lung zones.
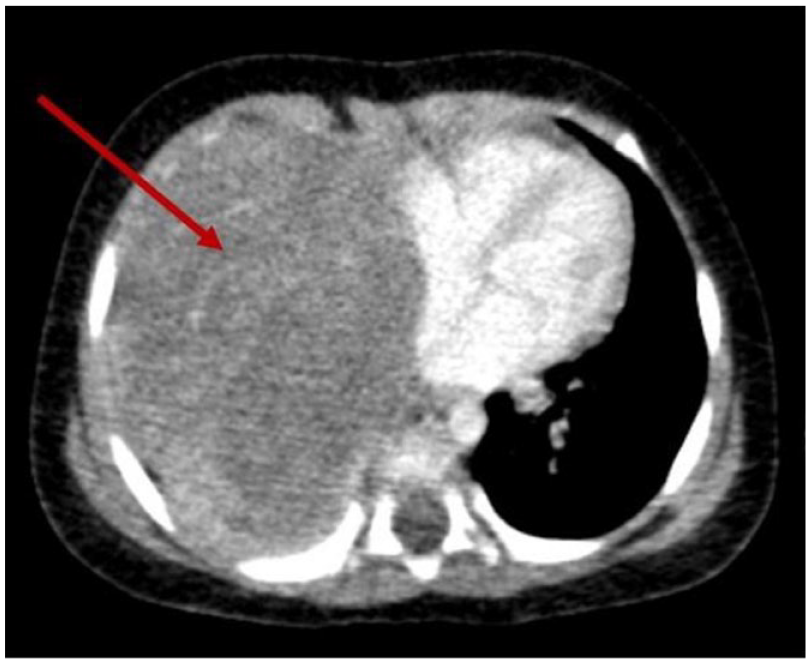
Contrast-enhanced CT of the chest showing large lesion with necrotic centre in the right hemi-thorax (arrow).
The infant underwent ultra-sound guided biopsy of the thoracic mass. Unfortunately, she deteriorated, developing a right-sided haemothorax and lung collapse. Despite intensive care and ventilation, she eventually succumbed. Histology report from the lung mass biopsy showed a highly cellular tumour similar to previous histology, likely a deposit from the previously resected mesoblastic nephroma (Figure 6).

Histological specimen – H&E staining of the lung mass biopsy specimen showing cellular proliferation and spindle cells suggestive of deposits from the CMN.
Discussion
CMN is an uncommon tumour arising from the renal mesenchymal tissue. It is mostly diagnosed during early infancy, and 15% of cases had been diagnosed during antenatal period. 7 It is usually an asymptomatic abdominal mass in the newborn detected incidentally. 2 However, it is also known to present with hypertension, haematuria, polyuria, and rarely as hypercalcaemia.5,8 Goldberg et al. 9 had reported an emergency presentation of CMN where an infant presented with haemoperitoneum and shock. This infant was diagnosed at 2 weeks of age, presenting with abdominal distension, haematuria and hypertension.
The main differential diagnoses of CMN are Wilms tumour and other rare renal tumours, such as clear cell sarcomas, which have an entirely different management and prognosis. Clinical symptoms, signs and imaging show similarity between those tumours, particularly with the cellular type of CMN. In comparison to a majority of CMN, only less than 2% of patients with Wilms tumour present during first 3 months of age. 2 Other differentiating features include Wilms tumour being associated with congenital syndromes, the predilection for presenting as bilateral tumours and the history of nephroblastomatosis. 5 The patient presented at 2 weeks of life as a unilateral tumour with no evidence of other congenital abnormalities, thus favouring a clinical diagnosis of CMN over Wilms tumour.
The standard treatment option for stage I and II CMN (80% of patients) is primary surgery. However, stage III (classic and mixed subtypes) CMN needs radical nephrectomy with adjuvant chemotherapy. 10 Based on these accepted recommendations, this patient underwent surgery without chemotherapy as she had a stage I tumour.
Although CMN is generally regarded a tumour with favourable clinical behaviour, recurrences and distant metastases have been reported in 5%–10% of cases. 11 However, only 1% of stage I/II tumours have been reported to recur, with the majority (75%) being local recurrences. 7 The first reported instance of clinically malignant behaviour of CMN was described in 1973 by Joshi et al., 11 where the tumour had infiltrated the ureter with multiple recurrences. 11 Reported cases indicate that metastatic and recurrent tumours are managed ideally with surgery and, when inoperable, resort to radio chemotherapy. Targeted therapy can be initiated in refractory cases. 12 The tyrosine kinase pathway is investigated in view of developing a more effective treatment for metastatic CMN. 13
Cases of metastatic CMN have reported liver, lung, brain, bone and heart metastases. Of the reported sites of metastasis, the commonest sites were the lung and liver. 12 Factors associated with increased likelihood of relapse were stage III disease, cellular type histology and diagnosis at 3 months of age or later. Patients who later developed relapses seemed to be initially diagnosed at a slightly older age (median age 3.8 months) compared to patients without relapses (median age <1 month). 14 Majority of relapses have occurred within 12 months after initial diagnosis, indicating that close monitoring of all cases should be advised for at least 1 year following initial treatment. 7 Albeit recurrences being rare, mortality among children with recurrences and metastasis is around 50%. There does not seem to be a difference in outcome among the patients who underwent surgery alone compared to those who received surgery with chemotherapy. 12
This infant had stage I disease, diagnosed at a very young age of 2 weeks with complete resection of the tumour at 1 month with tumour-free margins, no lymph node deposits and good recovery from surgery. The only factor that was suggestive of aggressive behaviour was the cellular, histological subtype.
Considering the molecular subtypes, CMN can be classified into NTRK3 (Neurotrophin TRK) fusion type, EGFR activation type and others. 15 The EGFR internal tandem duplication had been detected in next-generation sequencing assay in our patient. It is an intragenic and in-frame duplication. Alteration of the exon 18-exon 25 internal tandem duplication is described in CMN.16,17 This duplication involves exons 18–25 of the EGFR gene, which encode the entire EGFR tyrosine kinase domain. 16
Due to the rarity of the tumour, no local guidelines were available on follow-up. Abdominal ultrasonography was performed every 6 weeks along with systemic examination based on the premise that despite recurrences being rare, most were local recurrences.
The overall survival of patients with CMN is excellent; however, reported causes of death in more than half of the cases were treatment-related, and most of these patients were very young (median age, <1 year) as in this patient. 7 Treatment-related complications (both surgery and chemotherapy-related) seemed to be relatively high, especially when the tumour appeared in the first few weeks of life. Most deceased CMN patients were relatively young (newborns), and the main reason for death had been due to the inability to tolerate treatment-related complications. 7
Conclusion
This case report underscores the importance of being aware that CMNs can demonstrate malignant behaviour. Children may present with metastasis or recurrences later, even after complete excision. Therefore, appropriate follow-up and meticulous monitoring is necessary for early recognition. Further studies are needed to identify the patients who are at increased risk of developing recurrences and the optimal management of such patients.
Footnotes
Acknowledgements
Not applicable.
Author contributions
ASA, KWSMW, MS and VPW participated in management of the patient and providing clinical care. SAG and VJM contributed to the histological diagnosis, while KTEC contributed to immunohistochemistry and molecular genetics. ASA, KWSMW, MS, VPW, VJM and SAG contributed to writing up and editing of the article. All authors read and approved the case report prior to submission.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics approval
Our institution does not require ethical approval for reporting individual cases or case series.
Informed consent
Written informed consent was obtained from a legally authorised representative(s) for anonymized patient information to be published in this article.