
Abstract
An 18-year-old female with a history of atopic march, hyperhidrosis, and eosinophilic esophagitis was diagnosed with erythromelalgia and gastrointestinal dysautonomia secondary to presumed autoimmune small fiber neuropathy. The patient experienced significant clinical improvements after the initiation of intravenous immunoglobulin therapy, supporting an underlying autoimmune disorder.
Introduction
Erythromelalgia is a rare neurovascular disorder that presents with erythema, increased skin temperature, and peripheral limb pain. It can stem from primary microvascular or autonomic nervous dysfunction. Secondary associations include myeloproliferative disorders, connective tissue diseases, and vasculitis. 1 The pathophysiology of erythromelalgia may involve hypoxia, platelet aggregation, vascular abnormalities, endothelial edema, elevated temperature, prostaglandin release, and in some cases, small-fiber neuropathy (SFN) has been observed.2,3 Patients with primary erythromelalgia exhibit genetic alterations in the SCN9A gene, that encodes the Nav1.7 voltage-gated sodium channel predominantly expressed at nociceptive nerve terminals. 4
This case report discusses the diagnostic process and therapeutic approach for an 18-year-old female patient presenting with new onset erythromelalgia symptoms with gastrointestinal (GI) dysautonomia. These episodes were characterized by severe esophageal spasms, epigastric pain, gastroparesis, and nausea accompanied by chest pain, hypertension, and sinus tachycardia. 5 This patient had a complex medical history of gastroesophageal reflux disease (GERD), chronic constipation, eosinophilic esophagitis (EoE), atopic march, and hyperhidrosis.
The diagnostic process involved clinical evaluation, skin biopsy, laboratory tests, and imaging studies. Treatment strategies included analgesics, beta-blockers, antiepileptics, anti-inflammatory medications, and non-pharmacological approaches.1,6–8 This study emphasizes the need for detailed diagnosis and tailored treatment due to its impact on patients’ quality of life (QoL).
Case report
At age 18, the female presented to the dermatologist with new generalized flushing and bilateral extremity erythema and burning (Figure 1). Her medical history highlighted atopic march, childhood asthma, grade 2 pustular acne, and hyperhidrosis. At age 10, significant dysphagia led to an endoscopy, confirming EoE. After testing positive for milk protein allergy, she maintained a dairy-free diet. Although on proton pump inhibitor treatment, she continued to experience dysphagia and GERD episodes. She received budesonide treatments biannually. Endoscopies at ages 13, 15, and 17 indicated eosinophil counts consistent with GERD. In addition, an X-ray for chronic constipation incidentally diagnosed spina bifida occulta.

Photographic depiction of the patient’s lower extremities demonstrating erythematous manifestations on the feet, with notable involvement of the toes, consistent with erythromelalgia.
After a suspected Streptococcal infection at 17, the patient faced deteriorating GI symptoms. An upper GI endoscopy revealed a micro-perforation, confirmed by computed tomography (CT), leading to an emergency room visit for chest pain. After 5 days on antibiotics, the perforation healed, but she developed persistent esophageal spasms within 2 months. Progressively, distal-to-proximal erythematous flushing and burning pain emerged in the limbs and face, accompanied by chest tightness, epigastric pain, and hypertensive tachycardia.
Calcium channel blockers nifedipine and diltiazem were trialed for esophageal spasms but exacerbated face and limb flushing with paresthesia. She experienced hypertension, palpitations, tachycardia, GERD, epigastric pain, and esophageal spasms. 9 Episodic triggers included bowel movements, full bladder, food intake, showers of any temperature, and mild exertion. Some episodes, notably during sleep, lacked identifiable triggers. Traditional cooling methods, like ice baths, yielded limited relief.
Over time, a multidisciplinary team explored various therapies due to her persistent episodes affecting QoL. For her erythromelalgia, clonidine, sertraline and a topical compound of 5% ketamine, amitriptyline, and lidocaine provided some relief, while many treatments remained ineffective or had side effects. GI symptoms were managed with lansoprazole, mirtazapine, and metoclopramide. Acute episodes utilized Pro re nata (as needed) medications such as nitro spray and ondansetron.
In response to suspected autonomic dysfunction, the patient underwent a comprehensive investigation (Figure 2). Additional testing included Holter monitoring, stress test, echocardiogram, manometry, MRI, and CT scans. Testing eliminated postural orthostatic tachycardia syndrome, Jackhammer esophagus, paragangliomas, neuroendocrine tumors, pheochromocytoma, porphyria, and mast cell disorders, confirmed by molecular KIT mutation analysis and normal tryptase levels.
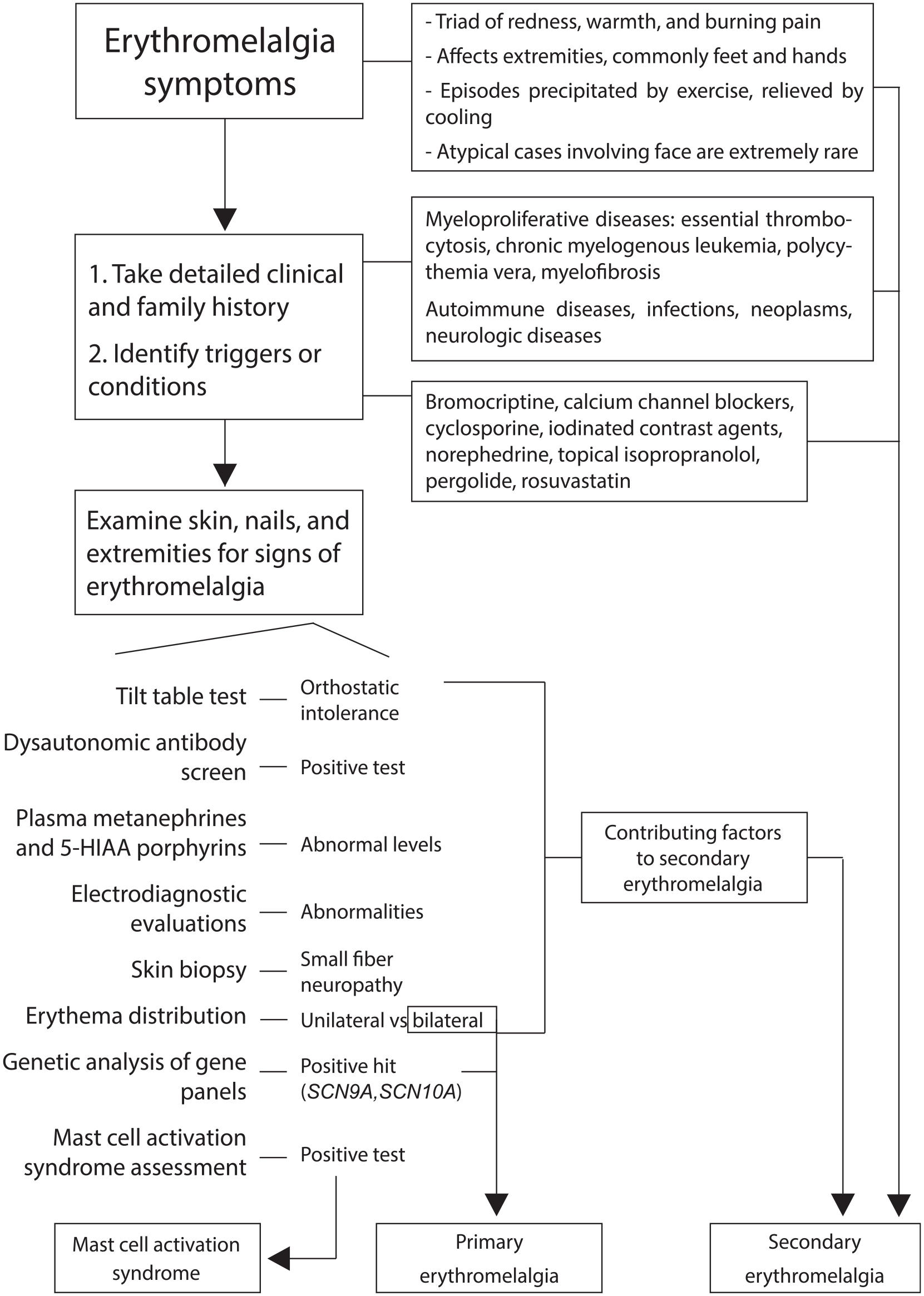
Diagnostic algorithm for non-infectious, red, and painful feet suspicious of erythromelalgia.
The genetic analysis, targeting genes associated with episodic pain syndromes and Charcot-Marie-Tooth neuropathy, found no significant markers for erythromelalgia or GI dysautonomia. In addition, standard Immunoglobulin G, Immunoglobulin A, and Immunoglobulin M levels, negative nuclear antibodies, rheumatoid factor tests, and a normal skin biopsy excluded lupus and other erythematous vascular skin conditions.
The skin biopsy showed a lower-end normal epidermal nerve density in the right calf (6.93) and a normal density in the right foot (7.85). Except for the nerve fiber test, all investigations were unremarkable, suggesting that the erythromelalgia and GI dysautonomia were likely secondary to an autoimmune SFN. The patient’s sensory and motor nerve conduction studies were normal.
Over 13 months, the patient underwent immunotherapy, starting with 55 g every 3 weeks of intravenous immunoglobulin (IVIG) that was increased to 60 g (due to 5 kg weight gain), leading to improved extremity temperature and GI symptoms but also side effects like migraines and nausea. This led to a dosage interval change to 20 g weekly.10–12 After 6 months of IVIG, side effects led to a transition to weekly 20 g subcutaneous immunoglobulin (SCIG) divided into 10 g twice a week. This regimen effectively managed erythromelalgia and GI symptoms and allowed for self-administration at home. The dosage was later optimized at 12 g twice a week, as increasing to 26 g caused side effects. After 8 months of immunotherapy, the patient was able to resume her university studies. Neurology advised tapering immunotherapy within 1–2 years if symptoms stabilized. The patient switched to levetiracetam (500 mg po BID), replacing cyproheptadine as its effectiveness diminished.
Discussion
Erythromelalgia poses diagnostic challenges and management complexities, which have significant physical and psychological implications for patients. Given the chronic pain and subsequent potential for psychological distress, prompt diagnosis, effective pain management, and mental health considerations are crucial for patient care. 13 This case report discusses the diagnostic challenges and therapeutic success in an 18-year-old female patient with erythromelalgia and GI dysautonomia, secondary to a putative autoimmune SFN. Although the specific autoantibody remains unidentified, the pronounced clinical amelioration following IVIG therapy would support an autoimmune etiology.
An illustrated flow diagram was created to depict the diagnostic approach to painful red legs, which was informed by a range of investigations (Figure 2). A tilt table test was employed to evaluate autonomic function, specifically in the context of potential dysautonomia. In this case, the outcome was within normal limits, signifying that the patient did not exhibit orthostatic intolerance or other variants of autonomic dysfunction that could contribute to her symptomatology. 14 We sought to detect anomalies in muscle structure or function indicative of a neuromuscular disorder. 15 Since the epidermal nerve density was at the lower threshold of the normal range, it is uncertain whether the patient’s symptoms were attributable to a structural abnormality. Mast cell activation syndrome (MCAS) testing was performed to assess a mast cell-mediated etiology. 16 Aberrant results could signify excessive mast cell activation, with flushing, pain, and GI perturbations; however, the test ruled out MCAS as a contributory factor.
The therapeutic management of this patient incorporated a blend of topical therapies, pharmacological interventions, and lifestyle modifications. First-line therapies including prostaglandins/prostacyclin or oral analogs were utilized with some success in symptom resolution. 17 Second-line therapies such as ketamine, amitriptyline, and lidocaine had moderate success.1,6–8,18 The patient’s erythromelalgia worsened with calcium channel blockers, contrary to its typical use as a third-line medication, consistent with previous reports. 19 There are a handful of case reports and cohort studies that report on the success of IVIG in secondary erythromelalgia.20,21
When standard treatments fail, IVIG can be considered due to potential autoimmune abnormalities in patients with secondary erythromelalgia. However, it is crucial to consider potential side effects, substantial costs, and resource limitations associated with this treatment.
Footnotes
Acknowledgements
The authors would like to acknowledge the diverse, multidisciplinary team from various hospitals involved in the patient’s diagnosis and continued care. This extensive collaboration included specialists from Pediatrics, Family Medicine, Gastroenterology, Neurogastroenterology, Dermatology, Genetics, Internal Medicine (with Dysautonomias focus), Hematology, Cardiology, Neurology, Immunology, Anesthesia and Pain Management, Emergency Medicine, Psychiatry, Pathology, Nursing, Homecare and Health Information Management.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article. R.G. Sibbald has participated in advisory boards for Profuse Medical and Novartis, has received honoraria from Medexus Pharmaceuticals, Inc. and Novartis, and is an investigator for a trial with Profuse Medical, Queen’s University, and ECHO Ontario.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Patient consent
The patient in this article has given informed consent to publication of their case details.