
Abstract
Cases of young patients combined with intracranial germinoma and parathyroid adenoma are extremely rare. A 6.25-year-old boy was diagnosed with growth hormone deficiency at his first visit and was then treated with growth hormone substitution. Later, he was clinically diagnosed with central diabetes insipidus (CDI) and primary hyperparathyroidism, whereas no abnormal imaging evidence was identified, except for a thickened pituitary stalk. Due to persistent follow-up, parathyroid adenoma and intracranial germinoma were verified in succession. The patient had derived benefits from parathyroidectomy and chemotherapy plus radiotherapy. We concluded that children and adolescents who present with CDI and pituitary stalk thickening should undergo repeated screenings for underlying intracranial germinoma. Multiple lesions involving the parathyroid gland and pituitary should alert physicians to the possibility of multiple endocrine neoplasia or other inherited diseases; therefore, genetic screening is recommended.
Keywords
Background
Intracranial germinomas are rare tumors that are often observed in very young patients. 1 Affected patients may present with obstructive hydrocephalus, nerve pathway or optic chiasm compression, endocrinopathy (most often diabetes insipidus), cognitive impairment, and mental status alterations. However, patients presenting with endocrine insufficiencies may have inconsistent visible magnetic resonance imaging (MRI) findings, causing a significant delay in diagnosis, 2 which emphasizes the necessity of persistent MRI screening. The diagnosis of intracranial germinomas in individuals is based on MRI, specific tumor markers, and tumor stereotactic biopsy. Currently, the standard treatment is a combination of chemotherapy (carboplatin/cisplatin and etoposide ± ifosfamide) and radiotherapy, followed by endocrine replacement therapy.3,4 Pure germinomas usually have a good prognosis, with 5-year and 10-year survival rates as high as 95.3% and 92.7%, respectively. 5 Primary hyperparathyroidism (PHPT) is a rare endocrine disorder in childhood that is characterized by hypercalcemia, elevated or normal parathyroid hormone (PTH) levels, and parathyroid lesions on imaging. By contrast, PHPT is relatively common in adulthood because of the popularization of routine measurement of serum calcium levels since the 1970s 6 and the introduction of osteoporosis screening guidelines targeting those with osteoporosis. Parathyroid adenoma is usually the primary cause of autonomous secretion of PTH in children. Parathyroidectomy is recommended as the only curative treatment for PHPT. 7 Here, we report our experience with a case of both intracranial germinoma and parathyroid adenoma and present a literature review of this condition.
Case presentation
A male patient aged 6 years and 3 months was admitted to our hospital for the first time due to an unsatisfactory height in October 2016. His birth weight and height were 3.0 kg (23rd percentile) and 50 cm (50th percentile), respectively. Further examination revealed a short (height 107 cm, <P3) and underweight (weight 15 kg, <P3) profile, growth velocity of less than 4 cm per year, low-level insulin-like growth factor-1 as 26.2 ng/ml (normal range: 52–419 ng/ml), delayed bone age, and negative brain MRI (Figure 1(a) and (b)). The arginine growth hormone (GH) test showed that the patient had complete GH deficiency for a peak GH level of 2.6 ng/ml and started GH substitution. With GH substitution, the growth velocity increased to 1 cm per month. However, this hormone replacement therapy lasted for 10 months and was terminated in October 2017 because the patient unexpectedly presented with polydipsia and polyuria.
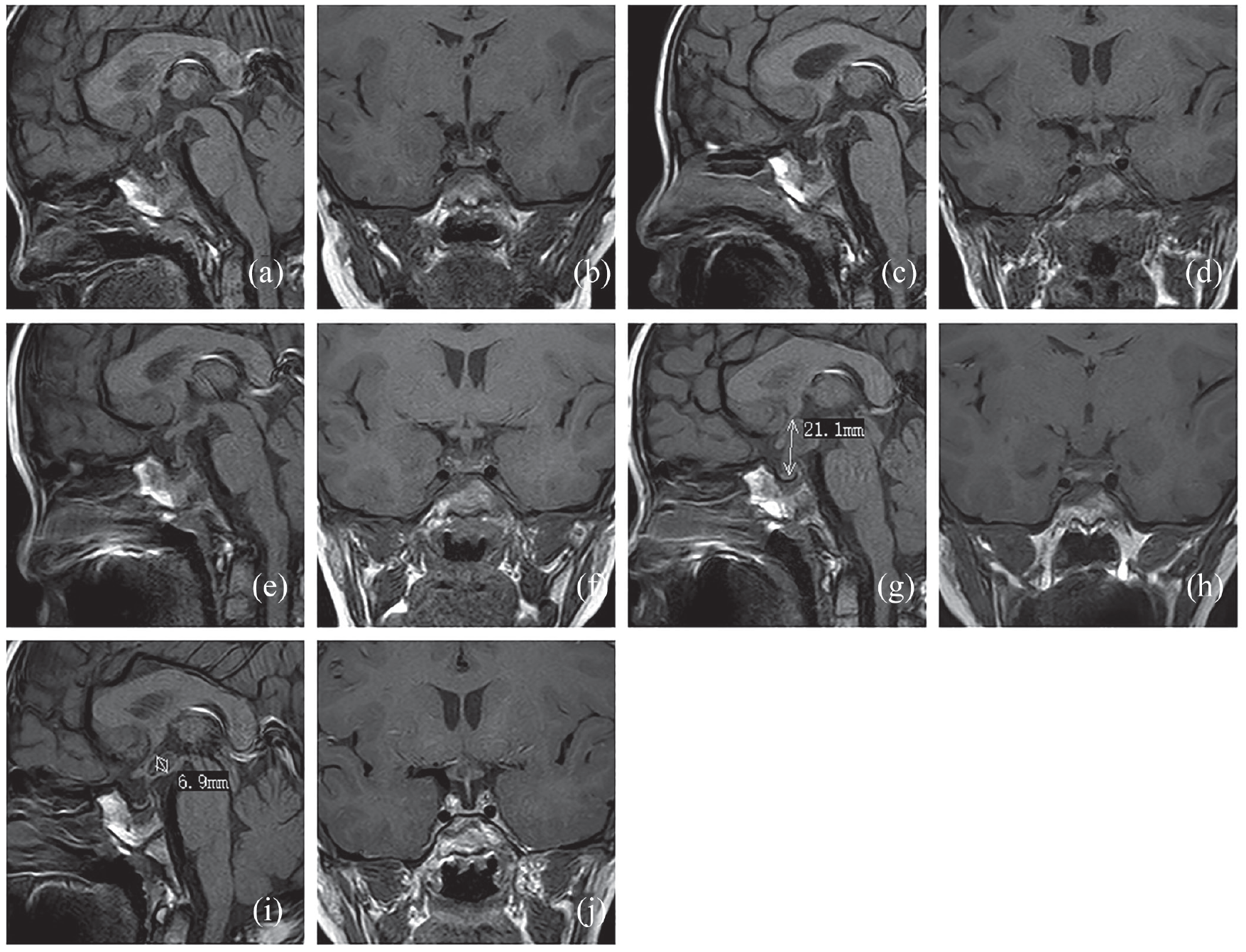
Magnetic resonance images of the pituitary gland.
The patient was again referred to our hospital and underwent a series of laboratory examinations. His total urine volume in 24 h was approximately 6200 ml. The specific gravity of the patient’s urine was 1.004 (normal range, >1.005). His serum sodium, potassium, glucose, and blood urea nitrogen levels were within normal ranges. During hospitalization, he suddenly experienced palpitations and vomiting. Persistent laboratory screening showed a high serum calcium level that met the diagnostic criteria for hypercalcinemia (Table 1). The emergence of hypercalcinemia confounded the classification of the etiology of diabetes insipidus (DI). Nevertheless, normal renal function, an ordinary urinary tract ultrasound, and most importantly, a high PTH level excluded a diagnosis of nephrogenic DI. The patient was evaluated using a water deprivation test followed by the administration of arginine vasopressin and was ultimately diagnosed with central DI (CDI). His serum β-human chorionic gonadotropin (hCG) and alpha-fetoprotein levels were normal, and his anterior pituitary hormonal profile was normal, except for GHD. The MRI of his brain and pituitary revealed a vanished posterior hypophyseal bright spot (PHBS) and a thickened pituitary stalk (TPS; Figure 1(c) and (d)). Furthermore, ultrasound and single-photon emission computed tomography of the parathyroid glands showed no positive findings. Based on the above evidence, the patient was simultaneously diagnosed with CDI and PHPT. A coincidence was that the two clinical diagnoses had no supportive evidence for imaging. Therefore, the optional treatment at that time was conservative. He was started on minirin to relieve polydipsia and polyuria, and miacalcic to control serum calcium. These drug therapies had a passable curative effect, but the patient had no increase in height. Regular follow-ups focusing on brain MRI, ultrasound of the parathyroid glands, and tumor markers were performed every 6 months. Interestingly, a mass in the upper-right position of the parathyroid glands was detected in August 2018, and the mass was excised immediately. The subsequent histological study suggested that the mass was a parathyroid adenoma. After surgical removal, the serum calcium and PTH levels returned to normal (Table 1), and the patient’s height started to increase at a growth velocity of 2.5 cm per year.
Treatments and serum analysis results from drug introduction until mass removal follow-up.

AFP: alpha-fetoprotein; Ca: calcium; normal values: 2.25–2.67 mmol/l; P: phosphorus; normal values: 1.45–2.1 mmol/l; PTH: parathyroid hormone; normal values: 15–68.3 pg/ml; W*: water deprivation test followed by the administration of arginine vasopressin; DEXA: dual-energy X-ray absorptiometry; GHD: growth hormone deficiency; Central DI: central diabetes insipidus.
As the patient still experienced polydipsia and polyuria, a routine brain MRI scan was continued. Consecutive brain MRI revealed a constant TPS (Figure 1(e) and (f)). As expected, a follow-up in February 2020 revealed a remarkably expanded pituitary stalk and a mass measuring 1.3 × 2.1 × 1.4 cm in the hypothalamus (Figure 1(g) and (h)). Meanwhile, an elevated serum β-hCG level was observed. Thus, the composite clinical data suggested a diagnosis of an intracranial tumor. The pathological diagnosis of a pure germinoma was confirmed based on a histological study of tumor stereotactic biopsy. The patient was treated with chemotherapy and sequential radiotherapy between May and September 2020. The treatment was effective and the patient did not develop polydipsia and polyuria with a small dose of minirin. The size of the hypothalamic lesion in recent images had reduced (Figure 1(i) and (j)). However, the adverse effect of the treatment arose as secondary hypothyroidism was noted with the need for Euthyrox replacement.
His parents denied a familial history of other neoplasia. Nevertheless, we obtained peripheral blood samples from the patient and his parents with informed consent and isolated genomic DNA to perform whole-exome gene detection. No mutation was detected.
The patient was presented at the clinic in July 2022 for his unsatisfied height. After active intracranial germinomas were ruled out, he was started on growth hormone treatment with a growth velocity >1 cm per month. Seven months later, his parents noted his breaking voice and sent him to the clinic again. This time we observed increased testicular volume (8 ml), rapid advancement of bone age (almost 3 years advance), high level of basal LH, and growth spurt, which all indicate that the boy was in rapidly progressive puberty. For fear of damaged adult height, he was started on GnRH analogs combined with growth hormone treatment. The case report was approved by the Ethics Committee of the West China Second University Hospital. The informed consent was obtained from the patient’s legal guardians.
Discussion
The initial symptom in our patient was diabetes insipidus (DI), a rare disease that can present at any age and can be classified on the basis of the damaged organs into central DI (CDI) and nephrogenic DI (NDI). 8 A water deprivation test followed by the administration of arginine vasopressin can be used to confirm the diagnosis of either CDI or NDI. 9 If CDI is confirmed, an MRI of the brain can help locate the areas of involvement, while evaluation of other pituitary hormone axes is recommended. CDI can be primary (most commonly septo-optic dysplasia, pituitary hypoplasia, holoprosencephaly, and some hereditary causes) or secondary (as a result of intracranial tumors, infiltrative disease, autoimmune hypophysitis, or injuries).10,11 The patient in our report exhibited invisible PHBS and a TPS at the initial MRI evaluation. Persistent follow-ups revealed progressive enlargement of the stalk and, ultimately, a mass lesion extending to the hypothalamus, which elicited a surgical biopsy with a pathological diagnosis of intracranial germinoma. The absence of the PHBS in the MRI of patients with CDI is common but nonspecific, and even a normal person could have identical changes. TPS can be detected in approximately one-third of children with CDI.12,13 Although the diagnostic criteria of TPS are controversial, some authorities consider that a pituitary stalk with a diameter >3 mm could be defined as TPS, and they further recommend that follow-up MRI scans are required every 3–6 months. 14 When the width of the pituitary stalk is greater than 7 mm or radiological thickening progresses to the third ventricle with further endocrinological deterioration, a surgical biopsy is necessary. 15 Germinomas and Langerhans’ cell histiocytosis (LCH) are the two most common underlying causes of CDI in pediatric patients. 16 Germinomas can secrete a small amount of β-hCG into the serum and cerebrospinal fluid, which can be used to differential between LCH and germinomas.
Intracranial germinomas are extremely rare, with a proportion ranging from 0.2% to 1.7% in intracranial tumors 17 and a predominant age of onset in childhood or adolescence. Germinomas arise mainly in the midline areas of the brain, particularly in the pineal and suprasellar regions. 18 Suprasellar germinomas in children usually present with hypothalamic/pituitary dysfunctions. The most common presentation is CDI followed by visual deficits, GHD, hypogonadism, and increased intracranial pressure. Take our patient for example, the pathological diagnosis of a pure germinoma was established by surgical biopsy 2.2 years after the onset of CDI. According to James Hayden et al., approximately one-third of the patients had prolonged symptomatology (>6 months) before definite diagnosis, and these cases are associated with a higher risk of metastatic disease. 19 Based on our case and a review of the literature, we recommend that patients with CDI and anterior pituitary hormone deficiencies should undergo follow-up MRI and specific tumor marker evaluation.
Notably, the patient in our report exhibited DI and hypercalcinemia simultaneously in the initial stage. Hypercalcinemia is also known to cause polyuria and polydipsia, which is classified as hypercalcemia-induced NDI. Further evaluation revealed an elevated serum PTH level in this patient, which was consistent with a diagnosis of PHPT. Term PHPT implies a high or inappropriately normal PTH level, resulting from excessive secretion from one or more parathyroid glands. Elevated PTH levels can trigger hypercalcemia, hypophosphatemia, and secondary organ-impaired symptoms characterized by skeletal (such as osteoporosis, ostalgia, bone fracture), renal (such as hypercalciuria and nephrolithiasis), gastrointestinal (pancreatitis and peptic ulcer disease), etc. 20 In our patient, the first parathyroid imaging was negative and did not meet the guidelines for parathyroidectomy; thus, we used miacalcic to lower the serum calcium level. However, the effect of the medication was disappointing. Repeat imaging of the parathyroid glands revealed a mass; thus, surgical resection was performed. The calcium level declined significantly thereafter, and the PTH levels normalized. This ideal effect verifies that parathyroidectomy remains the only cure for PHPT. 7 From Figure 2, we noticed that the growth velocity had stagnated during the phase of hypercalcemia and increased again after parathyroidectomy, which outlines the fact that bone resorption/destruction due to osteoclasts activated by elevated PTH severely blocked the height increase in our patient.
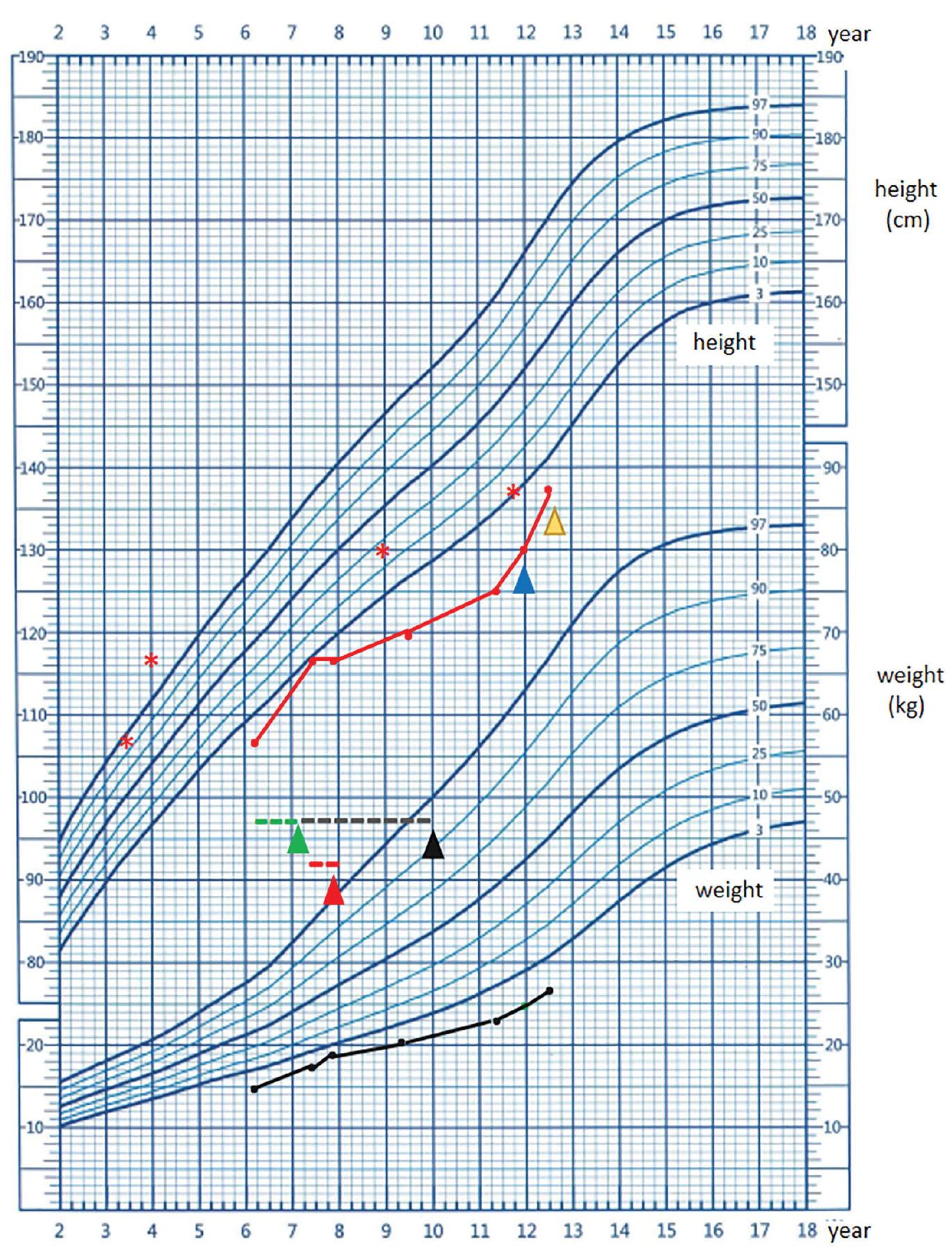
The red growth curve of our patient shows a meandering growth tendency.
The concomitant occurrence of solitary parathyroid adenoma and pituitary mass was suggestive of multiple endocrine neoplasia 1 (MEN1). MEN1 is a rare condition characterized by the occurrence of tumors involving the parathyroid glands, pancreatic islet cells, and the anterior pituitary. Parathyroid tumors occur in 95% of MEN1 cases with clinical manifestations of PHPT. 21 Anterior pituitary tumors occur in 30% of patients and manifest as either hyperactive adenomas (e.g., prolactinomas, somatotrophinomas, and corticotrophinomas) or non-functioning adenomas. 22 MEN1 is an autosomal dominant disorder with a germline MEN1 gene mutation. MEN1 mutational analysis is currently recommended for patients with parathyroid adenomas before the age of 30 years or for those with multi-gland parathyroid disease. 23
Although our patient had two sites of lesions involving the parathyroid gland and pituitary stalk, biochemical investigations revealed a growth hormone deficiency and a posterior pituitary hormone deficiency, which cannot meet the diagnostic criteria of MEN1. Further surgical biopsy revealed a solitary parathyroid adenoma combined with intracranial germinomas; most importantly, whole-exon genetic sequencing did not identify MEN1 mutation or any other pathogenic gene mutations. In addition, the curative effect in our patient was satisfactory, whereas patients with MEN syndrome had poor survival. On the whole, our patient was relieved of a suspicion of MEN1.
Conclusion
In summary, we report a rare case of combined intracranial germinoma and parathyroid adenoma. In patients with multiple endocrine tumors, MEN1 must be considered as a differential diagnosis; thus, genetic screening is recommended. Children and adolescents who present with CDI and TPS should be evaluated for intracranial germinomas. In our case, tumor markers were assessed and MRI scans were performed repeatedly until indicative evidence appeared.
Footnotes
Acknowledgements
We are grateful to the patient and his family who participated in this study. We also express our gratitude to all of the pediatricians, who provided the patient’s clinical data.
Author Contributions
Conceptualization, Jin Wu and Ying Liu.; Methodology, Tingting Zhang; Validation, Juanjuan Lv and Chuanjie Yuan; Formal analysis, Juanjuan Lv and Chuanjie Yuan; Investigation, Jin Wu.; Resources, Jin Wu; Data curation, Tingting Zhang.; Writing—original draft preparation, Tingting Zhang.; Writing—review and editing, Jin Wu and Ying Liu.; Visualization, Ying Liu.; Supervision, Jin Wu.; Project administration, Jin Wu.; Funding acquisition, Jin Wu. All authors have read and agreed to the published version of the manuscript.
Data Availability Statement
All datasets generated and analyzed for this study are included in the article/supplementary material.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics approval
Ethical approval to report this case was obtained from the Ethics Committee of the West China Second University Hospital. The ethics approval number is (2018) Clinical ethics approval No.4.
Informed consent
Written informed consent was obtained from a legally authorized representative(s) for anonymized patient information to be published in this article.