
Abstract
Hereditary palmoplantar keratoderma is a rare heterogenous group of genodermatoses characterised by hyperkeratosis of the palms and soles. Genetic alterations affecting proteins of the keratin cytoskeleton, cornified cell envelope, desmosomes and gap junction proteins have been implicated in the pathogenesis of inherited palmoplantar keratoderma. Reports of palmoplantar keratoderma in the African population are scarce. Herein, we report a case of a 29-year-old HIV-infected African female, who presented to a tertiary hospital with complaints of a painful left fourth toe, secondary to a constriction band. Her background history is significant for prior constriction bands involving her toes, some of which progressed to auto-amputations and childhood-onset thickening of the palmoplantar skin. Examination revealed diffuse transgrediens palmoplantar keratoderma with associated clinical findings of pseudo-ainhum and knuckle pads. A systemic workup was non-contributory. Next-generation sequencing genetic testing detected two variants of undetermined significance in gap junction protein beta 4, a connexin-encoding gene, and in the rhomboid 5 homolog 2 gene. Her phenotype remains discordant with our genetic findings. Her clinical features are instead consistent with overlapping phenotypes of gap junction protein beta 2-related connexin disorders: Vohwinkel syndrome and Bart–Pumphrey syndrome. Our case underlines the genetic heterogeneity of palmoplantar keratoderma and the diagnostic challenges it presents. Our patient required surgical amputation of the affected toe and is receiving ongoing dermatological management. Early recognition, appropriate referral and management are required to avert the debilitating consequences of mutilating keratoderma and improve the quality of life.
Introduction
Palmoplantar keratoderma (PPK) consists of a heterogeneous group of keratin disorders characterised by hyperkeratosis of the palms and soles resulting from an acquired or hereditary cause. 1 Acquired keratoderma is commonly a result of inflammatory dermatoses, systemic inflammatory disease, malignancy or infection, 2 while earlier age of onset, familial occurrence and presence of associated features allude to hereditary PPK. 1 While PPK may present as an isolated clinical finding, it may be associated with other cutaneous features or as a syndrome with extracutaneous manifestations in other organs. 3 Hereditary PPK are rare genodermatoses and the true global prevalence remains unknown. Limited studies report prevalence ranging as low as 4.4 per 100,000 in Northern Ireland, 4 to 3.1 per 10,000 in China, 5 and 5.2 per 10,000 in South India where consanguinity is common. 6 There is a dearth of literature concerning PPK among African populations.
Both ainhum and pseudo-ainhum are characterised by the formation of constriction bands involving any phalange which progresses to spontaneous auto-amputation. 7 Ainhum is idiopathic and typically involves the fifth toe. 8 It predominately occurs among tropical populations, having been reported in South America, the Indian subcontinent and Africa, where its prevalence ranges from 0.2% to 2%. 8 Pseudo-ainhum is characterised by constriction bands attributed to an identifiable cause which may be categorised as congenital, traumatic or secondary to disease. 7 Such disease may be hereditary such as the palmoplantar keratodermas. Non-hereditary causes of pseudo-ainhum include infections (leprosy, mycosis, syphilis), inflammatory dermatoses (psoriasis) and connective tissue disorders (systemic sclerosis, discoid lupus erythematosus). 7 Knuckle pads are thickened, skin-coloured plaques or nodules that overlie the small joints of the hand. 9
We present a case of childhood-onset PPK with associated features of pseudo-ainhum and knuckle pads in an African patient.
Case
A 29-year-old African female of Malawian ethnicity, an unemployed mother of two, presented to a tertiary hospital in August 2022 with complaints of a painful left fourth toe due to the formation of a circumferential constriction band.
The patient had experienced three prior occurrences of constriction bands involving her toes. The constriction bands first became apparent when she was 7 years old, involving her fifth toe bilaterally, which progressed to bilateral auto-amputations over a year. The next episode involved her right fourth toe at the age of 23 years for which she required surgical amputation for pain alleviation. As a result of the progressive loss of her toes, she experiences pain and mild functional limitations when standing for prolonged periods. She also reported skin changes which involved thickening of her palms and soles since early childhood. Other associated reported symptoms included Raynaud’s phenomenon 10 involving her fingers and joint stiffness involving the small joints of her hands. There were no complaints of auditory impairment, dental problems, dysphagia, muscle weakness, hyperhidrosis or sicca symptoms.
She was diagnosed with HIV infection in 2014 and is currently on a tenofovir, lamivudine and dolutegravir fixed-dose regimen but remains virologically unsuppressed with a viral load of 29,980 copies/mL, and a cluster of differentiation count (CD4) of 355 cells/µL. She has had no prior opportunistic infections. Her perinatal history is unremarkable, she was born to non-consanguineous parents and there is no history to suggest a collodion membrane at birth. No family history of similar skin disorders or cancers was reported. Presently, her children are not affected. She is a non-smoker with no prior significant occupational exposures or history of manual work.
Systemic examination was normal. Dermatological examination revealed diffuse, transgrediens palmoplantar hyperkeratosis with contiguous extension to the Achilles tendon and along the extensor tendon of the great toe (Figures 1 and 2). Skin-coloured hyperkeratotic plaques (knuckle pads) were observed over the dorsal surface of the metacarpophalangeal and proximal interphalangeal joints (Figure 3). A constriction band was present on the left fourth toe overlying the proximal interphalangeal joint (Figure 4). The neurovascular status of the left lower limb was intact. No xerosis, scaling or erythema of the skin was observed. Examination of her nails, hair, mucosa and dentition was normal.

(a) Diffuse palmar keratoderma and (b) diffuse plantar keratoderma.

Demonstrating transgrediens along the extensor tendon of the great toes.

Skin-coloured plaques (knuckle pads) overlying the metacarpophalangeal joint and proximal interphalangeal joints.

Constriction band involving the left fourth toe with a bulbous appearance of the distal portion of the affected toe.
Investigations were performed to exclude acquired causes due to the absence of a positive family history and features suggestive of systemic inflammatory disease. Routine laboratory tests were normal except for a microcytic hypochromic anaemia and low iron stores. Her erythrocyte sedimentation rate was elevated at 30 mm/h while her C-reactive protein remained normal. Her autoimmune, vasculitis and infectious screen was negative.
Radiographs of the feet revealed soft tissue alteration with no bony involvement (Figure 5). The patient did not consent to a skin biopsy. Over the years, her case has eluded diagnosis, despite presentations to multiple departments, including general surgery, orthopaedic surgery and internal medicine.

Radiograph of the left foot indicating a band-like constriction of the left fourth toe at the level of the proximal interphalangeal joint.
Genetic testing
The patient underwent genetic testing of 81 candidate genes commonly implicated in PPK in December 2022. Unfortunately, genetic analysis of her parents was not possible as they are both deceased. Cost and logistical limitations precluded the genetic testing of other first-degree relatives. Genomic DNA from a venous blood sample was enriched for targeted regions using a hybridisation-based protocol and underwent targeted next-generation sequencing (NGS) panel diagnostics using Illumina® technology (Illumina, San Diego, CA, USA). Two variants of uncertain significance (VUS) were identified (Table 1).
Results of genetic testing.

GJB4: gap junction protein beta 4; RHBDF2: rhomboid 5 homolog 2.
A heterozygous mutation in the gap junction beta 4 (GJB4) gene was detected c.653G>A (p.Gly218Asp). This missense mutation replaced glycine with aspartic acid at codon 218 of GJB4. This variant was found to be absent in the database of single nucleotide polymorphism (dbSNP, http://ncbi.nlm.nih.gov/SNP). An in silico analysis of the variant was found not to disrupt protein function. However, pathogenicity alone cannot be based on in silico models without functional studies, which in the context of connexin skin disorders is limited. 11 The absence of parental genetic analysis and out-of-family controls limited further bioinformatic analysis.
Gap junctions are intercellular channels that are formed when two connexon hemichannels align, which facilitate the transfer of metabolites and other small molecules. 11 Each connexon is composed of six connexin sub-units which may be of the same type (homomeric) or composed of different connexin types (heteromeric). 11 Connexins are expressed in multiple organs, including the cornea, cochlea, heart, peripheral nerves and skin. 11 At least nine different connexins (CX26, CX30, CX30.3, CX31, CX31.1, CX37, CX40, CX43 and CX45) are expressed in the skin in distinct or overlapping patterns. 11 The differential and co-expression of certain connexins are thought to play a role in keratinocyte differentiation, proliferation and repair. Connexin mutations involved in skin disorders are summarised in Table 2.12–14
Connexin-related skin disorders.
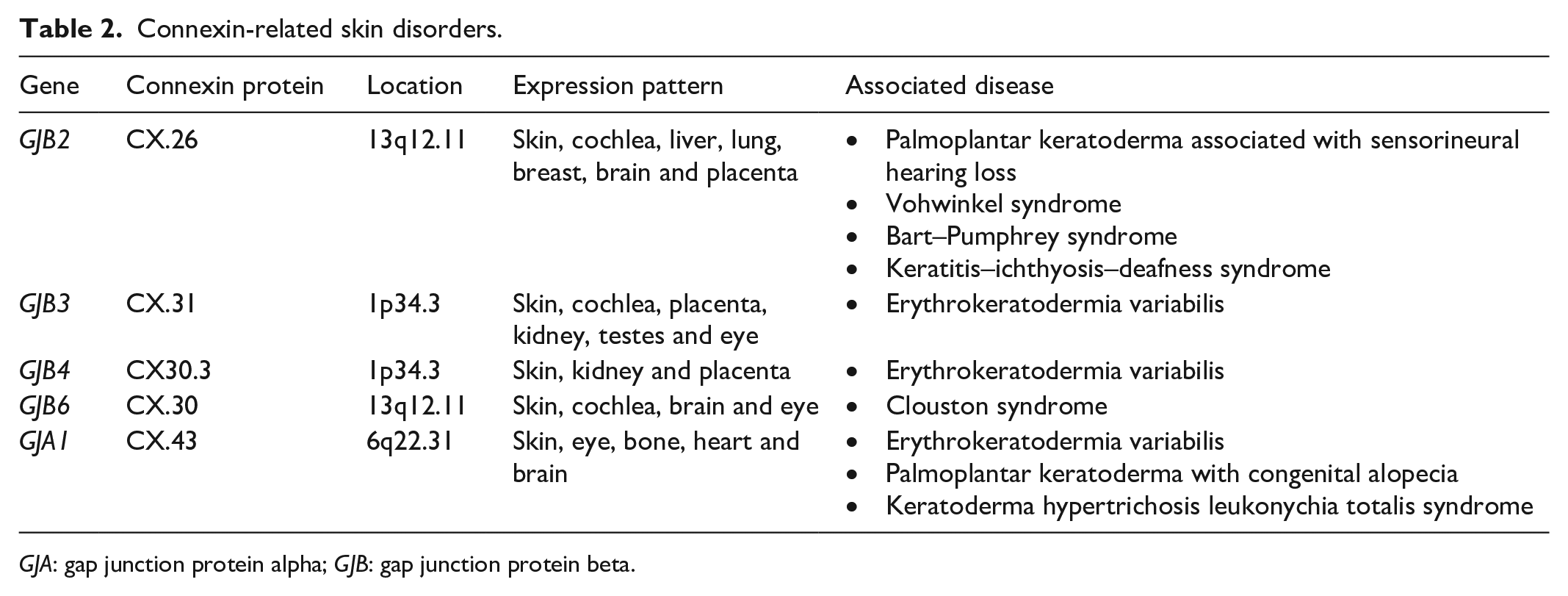
GJA: gap junction protein alpha; GJB: gap junction protein beta.
We describe a mutation in GJB4 encoding connexin 30.3 which is a beta connexin that is usually expressed in the stratum granulosum of the epidermis. 11 All beta-type connexins contain the amino acid glycine at position 12 (G12) which endows the protein with conformational flexibility. 15 Substitutions of glycine as reported in our case may have resultant structural and functional alterations in gap junction activity. Substitutions at G12 in two other beta-connexins (CX26 and CX31) have been established to result in skin disorders: keratitis–ichthyosis–deafness syndrome and erythrokeratodermia variabilis, respectively. 15 To the best of our knowledge, this presentation is the first to describe a GJB4 mutation associated with mutilating keratoderma. While in vitro functional studies and transgenic mice models have provided insight into connexin function, it remains unclear how mutant connexins may lead to pathophysiological changes contributing to the clinical phenotype in our case. Postulated mechanisms of pathogenesis include loss of gap junction function, abnormal gap junction function, mistrafficking defects and a dominant negative effect of mutant connexin on other connexin proteins. 14 Calcium is a regulator of keratinocyte differentiation; under homeostasis, higher levels are found in the stratum granulosum, with a markedly reduced amount in the outermost stratum corneum. 16 Thus alterations imposed upon by mutant connexins disrupt the epidermal calcium gradient affecting keratinocyte differentiation. 16 Further research is required to determine the precise pathways involved in the pathogenic processes in vivo especially as new variants are revealed.
Another heterozygous missense mutation in the rhomboid 5 homolog 2 (RHBDF2) gene c.581G>A (p.Arg194Gln) was observed in which arginine was replaced by glutamine at codon 194. RHBDF2 is the causative gene implicated in Howel-Evans syndrome, also known as tylosis with oesophageal cancer. This disease is characterised by PPK, oral precursor lesions and risk of squamous oesophageal cancer. 17 Mutations in RHBDF2, which encodes a rhomboid protease, result in dysregulated epidermal growth factor regulation, which has been implicated in the oncogenic process. 17 In silico computational bioinformatic scores predicting the RHBDF2 encoding rhomboid protein functionality are discordant in our case. While Sorting Intolerant from Tolerant reports the change as ‘tolerated’, Polymorphism Phenotyping version 2 (PolyPhen-2) categorised it as ‘potentially damaging’ which may have implications for follow-up and screening.
These mutations have been reported in the population genome aggregate database (GnomAD, https://gnomad.broadinstitute.org/) at 0.02% and 0.006%, respectively with no previous reports in the literature.
Follow-up and outcome
The patient underwent surgical amputation of the left fourth toe in September 2022. She was referred to dermatology for continued management. Emollients and topical keratolytic agents (urea, salicylic acid) were commenced. She began oral retinoids (acitretin) at a dose of 25mg daily. Three months later, she reports mild improvements. No new constriction bands were observed.
Discussion
Hereditary PPK can be classified according to phenotype into three broad categories: isolated PPK, PPK with distinctive cutaneous features and syndromic PPK with extracutaneous features. 3 It may be further differentiated by morphology and the extent of involvement as diffuse, focal or punctate.1,3 The nosology of PPK is evolving with advances in molecular genetic testing allowing for classification according to the causal gene defect.
Differential diagnoses
An approach to diagnosis based on morphology, the presence of transgrediens and features of pseudo-ainhum and knuckle pads yielded the following differential diagnoses among the known mutilating keratodermas: Vohwinkel syndrome, Bart–Pumphrey syndrome, loricrin keratoderma and Mal de Meleda. However, formal genetic testing did not reveal any pathogenic mutations in the causative genes.
Our patient presented with overlapping clinical features observed in some connexin disorders: Vohwinkel syndrome and Bart–Pumphrey syndrome. Vohwinkel syndrome is an autosomal dominant disorder that results from a mutation in the gap junction beta 2 (GJB2) gene located on chromosome 13q11-12, which encodes the protein connexin 26 (CX26). 1 It is characterised by a triad of diffuse PPK, starfish keratoses and pseudo-ainhum. 1 It has been closely associated with hearing loss in many cases. However, a formal audiological assessment did not reveal any hearing impairment in our patient. Similar presentations without hearing loss have been reported in families from Pakistan, 18 Iran 19 and in a case from India. 20 It has been reported sparingly in the literature and none reported in the African population. Another mutilating PPK with defects in GJB2 is Bart–Pumphrey syndrome which is characterised by leukonychia, knuckle pads and deafness. 21
While molecular analysis did not reveal a pathogenic mutation in GJB2, the patient had a missense mutation in another connexin-encoding gene, GJB4 (CX30.3), which is associated with erythrokeratodermia variabilis (EKV). EKV is a clinical manifestation characterised by hyperkeratotic plaques and transient erythematous areas, 22 a phenotype not consistent with our described presentation. Interestingly, Zhou et al. reported a Chinese patient with EKV and pseudo-ainhum but found no mutations in the putative causative genes GJB3 and GJB4. 23 Our patient’s clinical phenotype resembles an overlap of conditions caused by mutations in GJB2. Overlapping of phenotypes among connexin mutations has been demonstrated by Nemoto-Hasebe et al., who reported a Japanese case of diffuse PPK, pseudo-ainhum and sensorineural hearing loss consistent with classical Vohwinkel syndrome along with knuckle pads, which is a predominant finding in Bart–Pumphrey syndrome. 24 This patient was found to harbour a heterozygous missense mutation in GJB6 encoding CX30 instead, 24 and is yet another example of genetic heterogeneity.
Loricrin keratoderma or Camisa syndrome is the ichthyosiform variant of Vohwinkel syndrome resulting from a mutation in the LOR gene on chromosome 1q21 that encodes loricrin, a key feature of the cornified envelope which is needed to maintain the integrity of the stratum corneum. 25 These patients present with dry, scaly skin (ichthyosis) and maintain the classical features of Vohwinkel syndrome but without hearing loss. Our patient, however, displayed no features of ichthyosis. Rhambia et al. described an Indian case of classical Vohwinkel syndrome with sensorineural hearing loss as well as ichthyosis. 26 It is an interesting example of the variation of phenotypes, albeit unfortunate that no genetic testing was available.
Mal de Meleda is an autosomal recessive disorder with mutations in the SLURP-1 gene resulting in dysregulated keratinocyte apoptosis and consequent hyperkeratosis. 27 The phenotype includes diffuse transgrediens PPK, knuckle pads, pseudo-ainhum and other features such as nail dystrophies, hyperkeratotic plaques over joints and peri-oral erythema which were absent in our patient. 27 Lastly, the absence of peri-orofacial keratotic plaques, hair abnormalities and nail dystrophies makes Olmsted syndrome unlikely. 28
Our search of the literature revealed sparse reports of PPK in African populations with a Fitzpatrick phototype IV to V (Table 3).29–35 While PPK has been reported together with pseudo-ainhum and knuckle pads in some cases, to the best of our knowledge, we are the first to offer a genetic analysis for this presentation.
Summary of cases describing palmoplantar keratoderma among African populations.
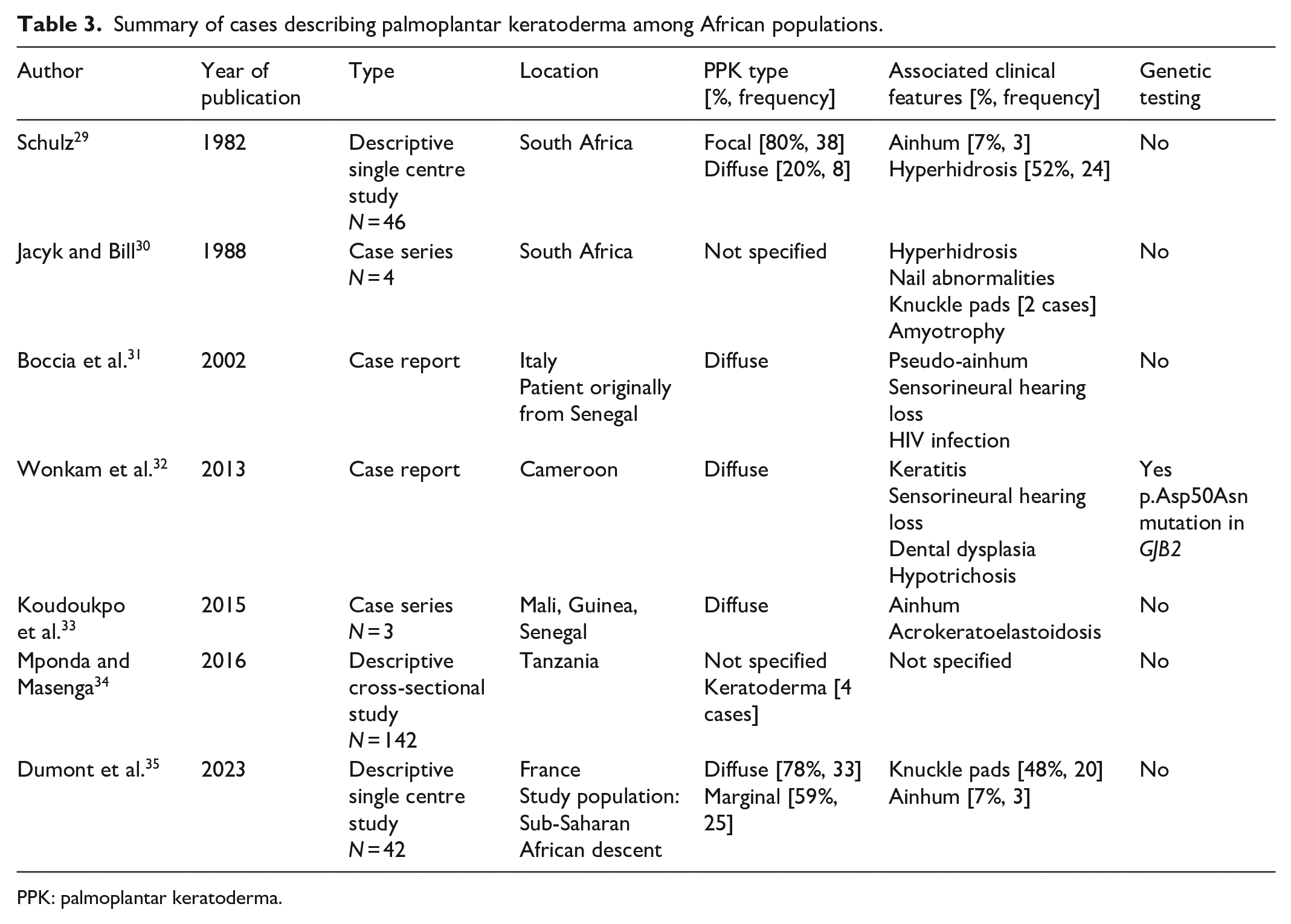
PPK: palmoplantar keratoderma.
Early identification of debilitating PPK is crucial for halting disease progression. In some cases, oral retinoids have been effective. 36 Conservative surgical techniques involving the excision of the constricting band and the use of Z-plasty flaps or skin grafts have been reported in the literature with varying degrees of success.37,38
Conclusion
Significant progress in the field of genomic medicine has yielded numerous genetic discoveries. Although genetic testing may be limited in certain contexts, the availability of NGS has enabled the identification of many VUS due to the high-throughput nature of this methodology. While the mutations described in this article are classified as VUS, they emphasize the need for more up-to-date population, disease databases and predictive algorithms to translate these findings into clinical utility. The genetic heterogeneity associated with PPK presents a diagnostic challenge, even with molecular testing, because there may be undiscovered PPK subtypes. Despite progress in identifying molecular gene defects, targeted gene therapies are not yet available. This case is presented for its unique characteristics and highlights the inadequate recognition of this clinical condition and the resulting consequences of delayed presentation in patients with mutilating keratoderma and resulting disabilities.
Footnotes
Acknowledgements
The authors thank the patient for the opportunity to share her medical condition for educational purposes.
Author contributions
The principal author (K.C.G.) made substantial contributions to the conception and design of the work, acquisition and interpretation of data, and writing of the article. The co-author (S.P.) made substantial contributions to the conception of the work, critically reviewing and editing the draft article. Both authors approved the final version of the article.
Data availability
All the data supporting our findings are contained within the article. Data sharing is not applicable to this article, as no new data were created or analysed in this study.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics approval
Our institution does not require ethical approval for reporting individual cases or case series.
Informed consent
Written informed consent was obtained from the patient for the publication of this case report containing anonymised patient information and accompanying images.