
Abstract
The coronavirus disease 2019 has been demonstrated to be a trigger for multiple immune-mediated diseases, such as antineutrophil cytoplasmic antibody-associated vasculitis. Associated vasculitis consists of rare autoimmune disorders that predominantly affect small vessels, leading to endothelial injury and tissue damage. We present a case of a newly diagnosed microscopic polyangiitis temporally associated with coronavirus disease 2019 infection in a previously healthy woman and a literature review. A 66-year-old female presented to the Emergency Room with fever, edema on her legs, productive cough, dyspnea, and hemoptysis. A chest computerized tomography scan revealed bilateral diffuse alveolar opacities with the appearance of diffuse alveolar hemorrhage. Blood analysis revealed a moderate normocytic, normochromic anemia with a hemoglobin of 6.6 g/dL, platelet count of 347 k/dL, leucocytes of 12,000/dL, a creatinine of 3.91 mg/dL (basal Cr: 0.9 mg/dL), and a Blood Urine Nnitrogen of 78 mg/dL. A urine sediment demonstrated glomerular hematuria, with mixed shapes of red blood cells. She was admitted to the intensive care unit and a bedside bronchoscopy revealed progressive bleeding with a bronchioalveolar lavage positive for diffuse alveolar hemorrhage. Given the critical involvement of the lungs and kidney function, the diagnostic approach revealed a positive p-anti-neutrophil cytoplasmic antibody on immunofluorescence and an anti-MPO (myeloperoxidase) level of 124.6 IU/mL. A renal biopsy demonstrated pauciimmune focal and segmental glomerulosclerosis. A diagnosis of microscopic polyangiitis triggered by severe acute respiratory syndrome coronavirus 2 infection was made, and immediate treatment with pulse-dose steroids and cyclophosphamide was initiated. The patient needed renal replacement therapy and was discharged for follow-up with nephrology and rheumatology services. The diagnostic approach of associated vasculitis can be more challenging in the coronavirus disease era. Atypical features in the pulmonary imaging and a rapid deterioration of the renal function should arise the clinical suspicion of the presence of an added condition to the coronavirus disease infection. Autoimmune conditions such as associated vasculitis should be evaluated even in the absence of previous autoimmune history. Prompt diagnosis and treatments must be prioritized to avoid end-organ definite damage. Further, larger and more collaborative studies are needed to confirm the potential role of coronavirus disease 2019 as a trigger of associated vasculitis.
Keywords
Introduction
The coronavirus disease 2019 (COVID-19) has proven to be much more than a pulmonary viral infection. In fact, COVID-19 has been demonstrated to be a potential trigger for multiple immune-mediated diseases, such as antineutrophil cytoplasmic antibody (ANCA)-associated vasculitides (AAV). 1 AAV is a group of rare autoimmune disorders that primarily affect small vessels leading to endothelial injury and tissue damage. This umbrella term includes granulomatosis with polyangiitis (GPA), microscopic polyangiitis (MPA), and eosinophilic GPA. 2 The classification of vasculitides based on the type of vessels involved and the underlying etiology was first established by the International Chapel Hill Convention Conference on the Nomenclature of Vasculitides (CHCC 2012). 3
MPA can involve small blood vessels in any organ or tissue but commonly affects the upper and lower respiratory tract, the kidneys, and peripheral nerves. 4 MPA is commonly associated with circulating MPO-ANCA, and clinical features include more severe renal disease and similar manifestations of GPA but without granulomatous inflammation. 4 The new MPA classification criteria include MPO antibodies positivity as an item that increases the odds for classification. 5 The incidence of MPA may vary across the globe, depending on multiple factors such as ancestry predominance of the population, or environmental factors like viral and bacterial infections.4,6
We present a case of a newly diagnosed MPA temporally associated with COVID-19 infection in a previously healthy woman and a literature review. For the reporting we utilized the framework proposed by Gasparyan et al. 7
Case presentation
A 66-year-old female with a family history that includes a mother with epilepsy, a father who died from hepatic cirrhosis, and a daughter who died because of chronic kidney disease of an unknown etiology. Her past medical history includes the following: 10 years of systemic arterial hypertension treated with angiotensin 2 receptor antagonists, osteoporosis diagnosed in 2007 and treated with bisphosphonates and dry eye diagnosed in 2019 and treated with lubricant eye drops. No previous history of autoimmune diseases was reported by the patient or in her family. She received two doses of the Pfizer BioNTech SARS-CoV2 vaccine in January and April 2022, respectively.
The patient’s current presentation began back in July 2022 with exertional dyspnea, which progressed to rest dyspnea over the next 2 weeks. She developed asthenia, fatigue, and orthopnea a week later. She was given ceftriaxone, ciprofloxacin, acetaminophen, diclofenac, ketorolac, omeprazole, and two intramuscular doses of iron dextran a month after the symptom’s onset. Nonetheless, the symptoms persisted, and she debuted on July 18th, 2022, with not measured fever, productive cough, and hemoptysis. As a result, she was taken to the emergency room, where a thoracic computerized tomography (CT) scan revealed bilateral diffuse alveolar opacities, a finding consistent with diffuse alveolar hemorrhage (DAH) (Figure 1(a)). On July 19th, 2022, before the patient was admitted to the ICU, a swab COVID test resulted positive as part of the admission protocol. Once in the ICU, a bedside bronchoscopy was performed, which confirmed the diagnosis of DAH after bronchoalveolar lavage.

(a) Axial thoracic CT displaying infiltrates with both lungs and diffuse, bilateral consolidative opacities with associated air bronchograms with sparing of subpleural space, (b) Puncture renal biopsy: ischemic type sclerosant glomeruli, mesangial expansion, segmentary necrotizing lesions with fibrin: compatible with focal and segmentary glomerulonephritis, and (c) direct immunofluorescence negative for Immunoglobulins G, A, M, Cq1, C3c, C4c, and fibrinogen.
At the time of her admission, her blood analysis revealed the following remarkable findings: a moderate normocytic normochromic anemia (hemoglobin 6.6 g/dL (Upper limit of normal: 12.5 mg/dL), hematocrit 19% (ULN: 30%), mean corpuscular volume 93.9 fL (normal: 80–100 fL)), platelets 347,000/dL (normal 100,000–450,000/dL), leucocytes 12.1 k/dL (normal: 4–11.5 k/dL), creatinine 3.81 mg/dL (normal 0.7–1.1 mg/dL), Prothrombin time: 12 s, INR: 1, and Partial thromboplastin time: 30 s (normal: 11–13.5 s, 0.8–1.1, 25–35 s, respectively). Serum potassium was 6.6 mEq/dL (ULN: 5.5 mEq/dL) An enzyme-linked immunosorbent assay (ELISA) for ANCA antibodies was performed, and anti-MPO autoantibody was found to be positive.
Following that, urinalysis sediment was performed, which revealed dysmorphic red blood cells. The estimated glomerular filtration rate abruptly declined in the following days, prompting a renal biopsy. The pathology reported active and fibrous extracapillary proliferative glomerulonephritis, diffuse pauciimmune type, with fibrinoid necrosis and segmental sclerosis, active tubulo- interstitial nephritis, active multifocal tubular injury, interstitial fibrosis grade II (40%), and moderate arteriosclerosis. The immunofluorescence demonstrated pauci-immune deposition of immune complexes (Figure 1(b) and (c)).
The patient was treated with three boluses of 250 mg of methylprednisolone and continued rapid prednisone tapering by the PEXIVAS protocol, and cyclophosphamide. Plasmapheresis was not attempted due to a good clinical response by the patient with the initial regimen. Hemodialysis was started, given the important and acute decrease in glomerular filtration rate and severe hyperkalemia. The patient was discharged after 2 weeks from the hospital with follow-up appointments at the Rheumatology and Nephrology clinic, remaining in permanent renal substitutive therapy. 4
Literature review
We performed a literature review according to the narrative biomedical review, 7 conducting a search strategy from December 1st, 2019 to October 31st, 2022, in Scopus and PubMed using the MeSH search terms (“Coronavirus” OR “COVID-19” OR “SARS-CoV-2” OR “Sever Acute Respiratory Syndrome Coronavirus 2” AND “Anti-Neutrophil Cytoplasmic Antibody-Associated Vasculitis” OR “ANCA”). All types of studies in English were included. We extracted the type of study, author, year of publication, demographics, previous history of autoimmune conditions, serological profile, renal pathology, renal replacement therapy, AAV treatments, and AAV sequels.
Discussion
We reported a case of new-onset MPA temporally associated with SARS-CoV-2 infection as a potential immune trigger that presented with the life-threatening pulmonary-renal syndrome in a previous healthy female.
Pulmonary involvement is very common in both COVID-19 infection and AAV. In COVID-19, the main radiological features are multifocal bilateral ground-glass opacities with a predominance of the peripheral and posterior zones of the lungs. While in AAV with DAH, a patchy ground-glass appearance or nonspecific interstitial pneumonia, with variability depending on the severity. Given this, in any patient with suspected autoimmune pulmonary diseases, COVID-19 should be ruled out, as in our case.
In our literature review, we found eight cases of new-onset AAV temporally associated with COVID-19 in adults (Table 1).2,8–12 After inclusion of the present case, out of the nine patients, five (55%) were female and the mean age was 49.77 years. One (11%) patient had scleroderma, and one (11%) patient had rheumatoid arthritis. The rest (78%) did not have any history of autoimmune disease. A c-ANCA report by immunofluorescence was the most common pattern of ANCA antibodies in five (55%) of the cases. Not all the cases reported an ELISA result for ANCA antibodies. Of the five cases that were reported, three (60%) were MPO-positive. In the renal biopsy, all the patients revealed suggestive findings of AAV, with pauciimmune deposition and crescentic glomerulonephritis, or glomerulosclerosis. All patients received pulse-dose steroids with oral steroid tapering. Five (55%) received cyclophosphamide, and four (45%) received rituximab as an induction regimen. As noted, three (33%) patients remained on permanent renal replacement therapy. One (11%) patient had permanent hearing loss, and five (55%) remained free of permanent sequels at the last follow-up. Even though COVID vaccination has also been linked to new-onset AAV, given the period after vaccination in our patient and the acuity of the COVID infection, clinically, the active infection was more probable to be associated with the AAV features.
Cases reported in the literature of adults with new-onset ANCA-associated vasculitis temporally associated with COVID-19 infection.
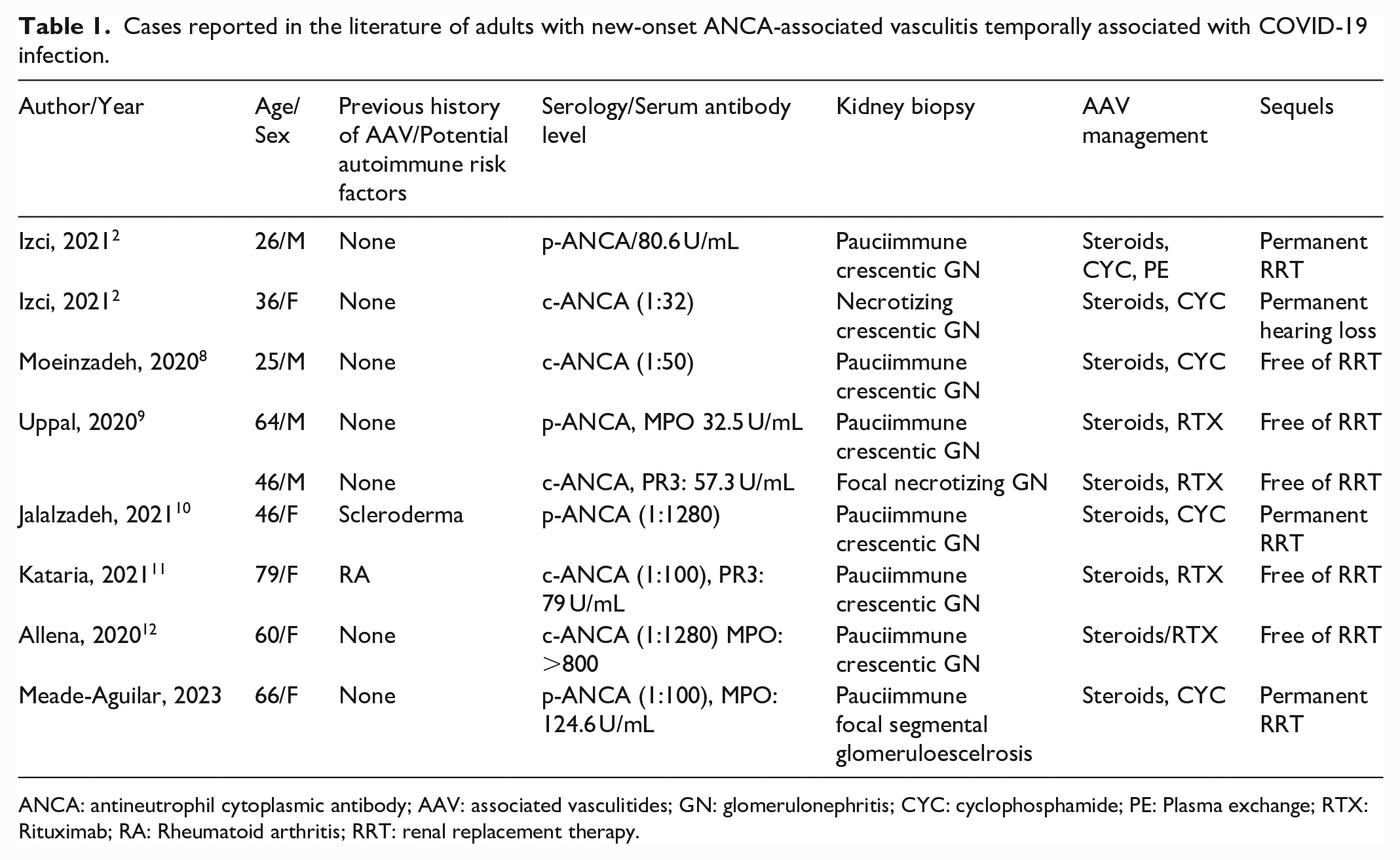
ANCA: antineutrophil cytoplasmic antibody; AAV: associated vasculitides; GN: glomerulonephritis; CYC: cyclophosphamide; PE: Plasma exchange; RTX: Rituximab; RA: Rheumatoid arthritis; RRT: renal replacement therapy.
Based on the new classification criteria for GPA and PMA, 5 from the included patients, four (44%) have a GPA phenotype, and the rest (56%) have a Microscopic polyangiitis phenotype. Data on AAV epidemiology in Latin America are scarce. A systematic review 13 of the epidemiology of AAV in the Hispanic population described a predominance of GPA over PMA in Mexico and Brazil. A Mexican retrospective cohort 14 showed that 9 of 10 patients with AAV have a GPA phenotype. Thus, the proportion of patients with GPA and MPA triggered by COVID-19 infection may follow the same proportion as the one presented by the population.
Severe COVID-19 infection is associated with a high rate of acute kidney injury. 15 Therefore, in the context of a newly diagnosed AAV or relapse and COVID-19, special attention to the urinary sediment should be paid. Active urinary sediment, the presence of subnephrotic range proteinuria, and the clinical and sudden decline in renal function should prompt AAV diagnostic approach.
One of the most relevant risk factors for developing autoimmunity is the presence of previous autoimmunity phenomena. 16 Nonetheless, given that most of the patients did not have any previous history of autoimmune diseases, patients without previous history of autoimmunity should also be assessed for AAV in the presence of consistent clinical manifestations, as previously mentioned.
Conclusions
Further, larger and collaborative studies are needed to confirm the potential role of COVID-19 as a trigger of AAV. The diagnostic approach of AAV can be more challenging in the COVID era. Atypical features in the pulmonary imaging and a rapid deterioration of the renal function should arise the clinical suspicion of the presence of an added condition to the COVID infection. Autoimmune conditions such as AAV should be evaluated even in the absence of previous autoimmune history. Prompt diagnosis and treatments must be prioritized to avoid end-organ definite damage.
Footnotes
Acknowledgements
None
Author contributions
JAMA, ORPN, JNVM, and SPRE contributed to conception of the idea, coordination, article preparation, and article review. ESH, GABA, TOML, EDT, and MPP contributed with data acquisition and conception of idea. All authors read and approved the final article.
Data availability statement
The data presented in this study are available on request from the corresponding author. The data are not publicly available due to privacy restrictions.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics approval
Ethical approval to report this case was obtained from Comite de Etica del Hospital General de San Juan del Rio, 001-1288568250-1.
Informed consent
Written informed consent was obtained from the patient(s) for their anonymized information to be published in this article.