
Abstract
A 62-year-old man was admitted to the emergency department with hypotension and altered consciousness. On physical examination, he had hyperpigmentation of the skin and mucous membranes. Admission tests revealed hypoglycemia, hyponatremia, and hyperkalemia. Fluid resuscitation was initiated with no improvement in blood pressure. Because adrenal crisis was suspected, blood samples for cortisol and adrenocorticotropic hormone were collected before commencing hydrocortisone, after which blood pressure improved and electrolyte disturbances disappeared. The tests revealed decreased serum cortisol and an increase in adrenocorticotropic hormone. A magnetic resonance imaging scan of the abdomen revealed evidence of bilateral adrenal hemorrhage. Positive antiphospholipid antibodies were detected during the investigations. This case underscores the importance of prompt evaluation of clinical signs and symptoms that may indicate adrenal crisis.
Introduction
Bilateral adrenal hemorrhage (BAH) is a rare phenomenon first described by Canton in 18631,2 that can have catastrophic consequences in the setting of adrenal crisis. It has been associated with several entities, notably infections in the context of Waterhouse–Friderichsen syndrome. 1 A rare etiology is antiphospholipid syndrome (APS). There are cases of BAH in patients taking anticoagulants because of previous thrombotic events or using anticoagulation as prophylaxis in the postoperative period who are later diagnosed with APS. However, very few cases of APS have been described as the initial manifestation of adrenal crisis secondary to adrenal hemorrhage. Fewer than 10 cases are currently described in the literature. 3 In cases of clinical suspicion, replacement therapy with hydrocortisone or methylprednisolone should be initiated promptly after baseline cortisol levels are determined, and confirmation by computed tomography (CAT) or contrast-enhanced magnetic resonance imaging (MRI) should be obtained. 4
Case
A 62-year-old man was admitted to the emergency department due to asthenia, adynamia, and diffuse abdominal pain of 1 week duration, without nausea, emesis, or changes in stool consistency. He had also lost about 5 kg of weight in 1 month. He denied having had this clinical picture before. He had no important personal or family history. On physical examination, he was hypotensive, blood pressure (BP) 80/50 mmHg, tachycardic heart rate (HR) 110 beats per minute (bpm), with altered state of consciousness Glasgow score 13/15, 5 without desaturation or fever. He had no signs of peritoneal irritation. He had brown pigmentation on the arms, forearms, back of the hands, legs, and oral mucosa (Figure 1). No other abnormalities were noted on physical or neurologic examination.

Increased brown pigmentation on the back of the hand (a), the lower alveolar mucosa in the oral cavity (b), and the tongue (c)
He was admitted to the resuscitation unit, where he received an intravenous (IV) fluid bolus of 20 cc/kg without improvement in BP. Initial paraclinical testing (Table 1) revealed anemia, hyponatremia, hyperkalemia, metabolic acidosis, and an increase in acute phase reactants. Given the physical examination findings, a possible adrenal crisis was suspected. Therefore, IV hydrocortisone (100 mg IV every 8 h) was administered after serum cortisol and adrenocorticotropic hormone (ACTH) were measured.
Paraclinical tests during hospitalization.
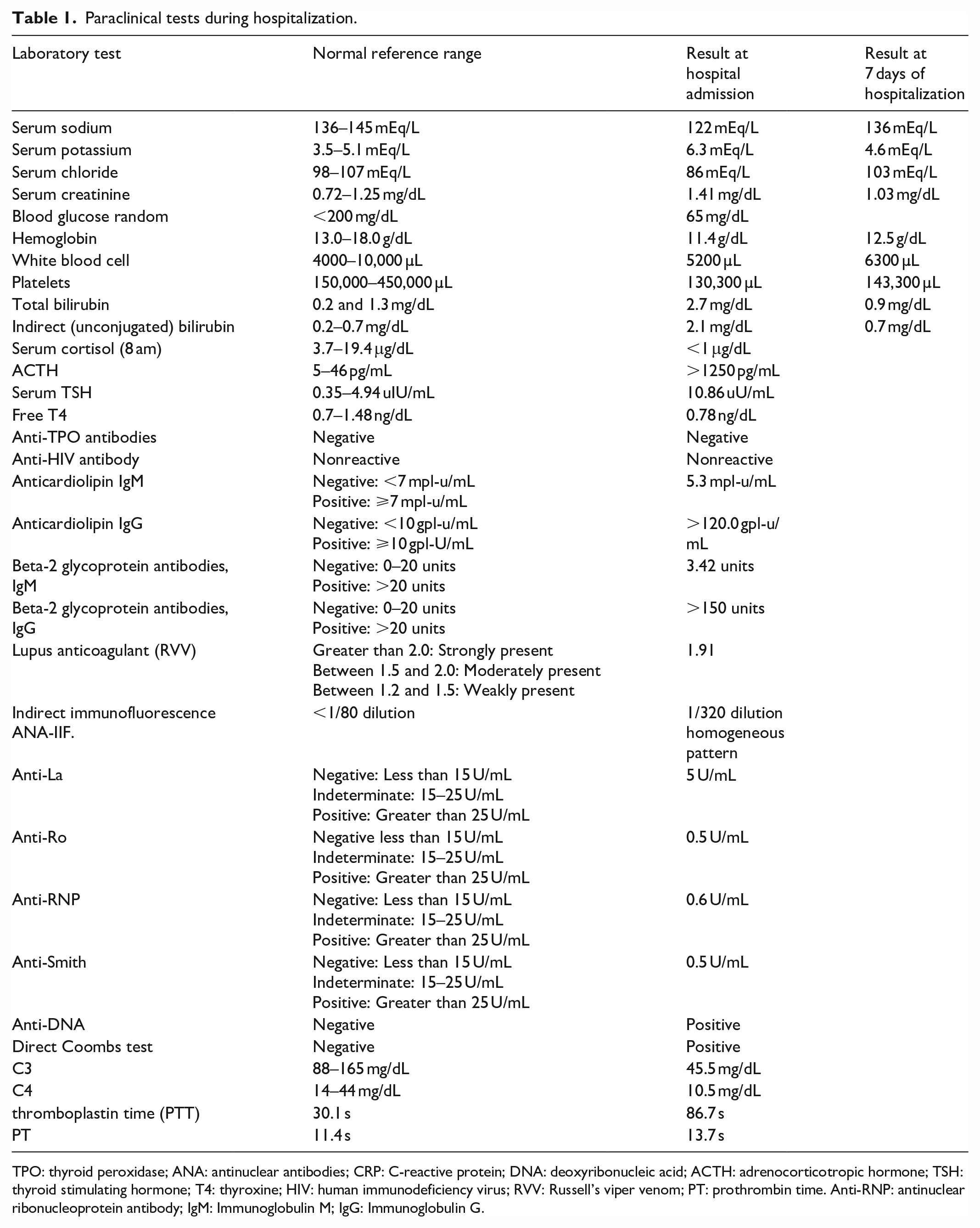
TPO: thyroid peroxidase; ANA: antinuclear antibodies; CRP: C-reactive protein; DNA: deoxyribonucleic acid; ACTH: adrenocorticotropic hormone; TSH: thyroid stimulating hormone; T4: thyroxine; HIV: human immunodeficiency virus; RVV: Russell’s viper venom; PT: prothrombin time. Anti-RNP: antinuclear ribonucleoprotein antibody; IgM: Immunoglobulin M; IgG: Immunoglobulin G.
Laboratory values were consistent with primary adrenal insufficiency. Specifically, serum cortisol was <1 μg/dL (3.7–19.4 μg/dL), and ACTH was >1250 pg/mL (5–46 pg/mL). In addition, subclinical primary hypothyroidism was detected. Therefore, after starting IV corticosteroids, substitution with levothyroxine at a dose of 1.6 mcg/kg was performed to avoid perpetuation of adrenal crisis.
An imaging study was performed with contrast-enhanced abdominopelvic tomography, which showed diffuse thickening and delayed enhancement of adrenal glands. The right adrenal gland showed hyperdense areas in the body corresponding to areas of subacute hemorrhage. Abdominal MRI with gadolinium contrast was also performed, which revealed lobulated adrenal glands with generalized thickening of the body and arms and a hyperintense adrenal lesion on both T1- and T2-weighted images. These findings were consistent with BAH in subacute stage (Figure 2).
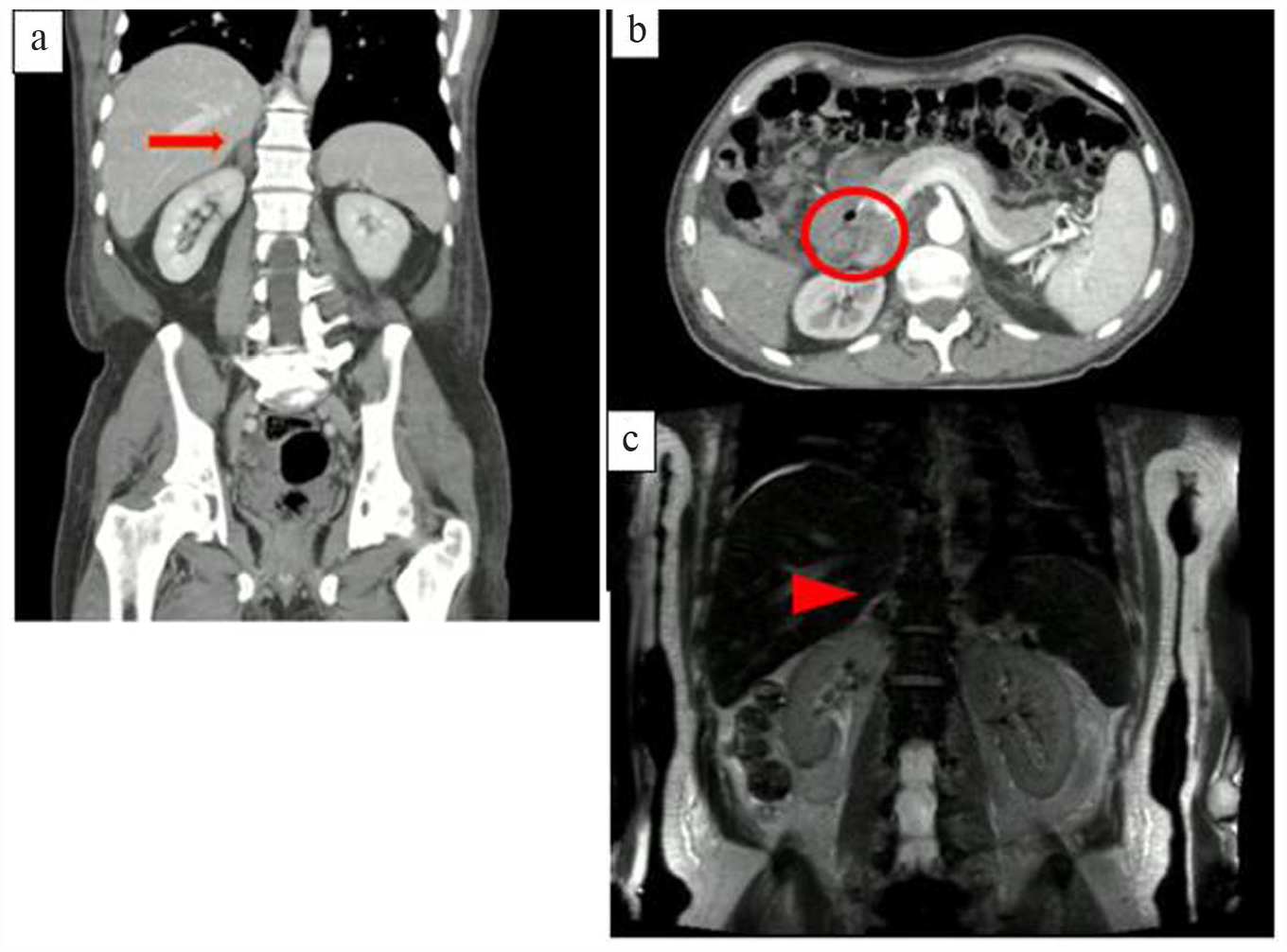
Contrast-enhanced abdominopelvic tomography (a) with evidence of adrenal glands with diffuse thickening and delayed enhancement after contrast administration (arrow). (b) The right adrenal gland shows hyperdense areas in the body corresponding to areas of subacute hemorrhage (circle). Abdominal MRI with gadolinium contrast (c) with evidence of lobulated adrenal glands with generalized thickening of the body and arms, with low signal intensity in the T2 sequence (arrowhead). Findings suggestive of bilateral adrenal hemorrhage in the subacute stage. MRI: magnetic resonance imaging.
Blood cultures, human immunodeficiency virus (HIV) serology, treponemal test, hepatitis B, hepatitis C, and bacilloscopies were performed as part of the investigations to search for etiology, all of which were negative (Table 1). Autoimmunity studies were extended with negative anti-SCL (DNA topoisomerase I). Coagulation proteins S and C and coagulation factor V were normal. In the studies looking for APS, positive lupus anticoagulant, anticardiolipin antibodies, and beta-2 glycoprotein were found, configuring a triple-positive APS.
Patient’s follow-up
After 3 days of treatment, clinical and paraclinical improvement was documented (Table 1) and the patient was transferred to the general ward. There he recorded normal BP, and the electrolyte disturbance was resolved. He was evaluated by the endocrinology service, which initiated a progressive steroid weaning down, and 7 days later, he was discharged with oral corticosteroids: Hydrocortisone 10 mg in the morning and 5 mg at night and fludrocortisone 100 μg/day.
In addition, laboratories detected antinuclear antibodies (ANA) above the laboratory reference range, positive anti-dsDNA antibodies, low complement levels, hemolytic anemia, leukopenia, and thrombocytopenia (Table 1). He was evaluated by the rheumatology service, which considered systemic lupus erythematosus (SLE) according to the Systemic Lupus International Collaborating Clinics (SLICC) classification criteria 6 and APS secondary to SLE. After confirming that no further bleeding was present, he was discharged on anticoagulant management with a target INR (international normalized ratio) of ⩾2.0, given the high risk of thrombosis due to the triple-positive APS. Considering the finding of SLE, treatment was, in addition, supplemented with azathioprine and hydroxychloroquine.
Control examinations were performed after 12 weeks (approximately 3 months) and switched from warfarin to enoxaparin prior to laboratory testing. Reports demonstrated anticardiolipin IgG, lupus anticoagulant, and beta-2 glycoprotein IgG antibodies persistently positive, meeting criteria for antiphospholipid antibodies (aPL) syndrome.
Discussion
Adrenal crisis is defined as a deficiency in the synthesis and secretion of steroid hormones, a life-threatening condition with mortality risk of 0.5/100 patients per year. 1 BAH is a rare phenomenon that can have catastrophic consequences in the setting of adrenal crisis. It has been associated with various entities, such as infections in the context of Waterhouse–Friderichsen syndrome,1,2 trauma, surgery, and infections caused by pseudomonas aeruginosa. 3 Primary adrenal insufficiency may also occur in the context of autoimmune polyendocrinopathy syndromes. However, adrenal hemorrhage is not common.
Adrenal hemorrhage secondary to APS is a rare complication observed in 0.4% of patients with APS and may be one of the first manifestations of this autoimmune disease. 3 Venous thrombus formation in the adrenal gland has been described, evolving into hemorrhage and/or venous infarction. Half of patients with this disease have a history of venous or arterial thrombosis, and 97% of cases have a positive lupus anticoagulant test. 4 In our case, the patient had APS secondary to SLE with triple positivity for aPL, which is considered a worse prognosis for thrombotic events and mortality. 4 Though endocrine abnormalities are not a common manifestation or complication of APS, adrenal insufficiency has been reported as the most common endocrine manifestation. 6
The pathogenesis of adrenal insufficiency in APS is not clearly understood. It is unrelated to autoantibody production and is essentially vascular. The unique nature of the vascular anatomy of the adrenal glands with an abundant arterial supply but limited venous drainage through a single vein may predispose patients to thrombosis. Results of the histopathologic study of adrenal glands showed that hemorrhagic infarction with venous thrombosis was the main finding. These data are considered evidence of adrenal vein thrombosis as the cause of adrenal hemorrhage. 7
Diagnosis of adrenal crisis is difficult because of nonspecific symptoms such as hypotension, hypovolemia, nausea, abdominal pain, fatigue, among others, for which paraclinical tests such as sodium, potassium, glucose, and serum cortisol must be performed. 8 Serum cortisol at 8 am is clinically useful to rule out adrenal insufficiency. Levels above 20 mg/dL (550 nmol/L) make the diagnosis of adrenal insufficiency unlikely, whereas a cortisol level of less than 5 mg/dL supports the diagnosis. 9 The adrenocorticotropic hormone stimulation test may be useful, although it is not required for initiation of treatment in adrenal crisis and has not been specifically validated in this context. 9
In the context of imaging studies, CAT of the abdomen and MRI are useful for diagnosis. 10 In acute hemorrhage, tomography shows an enlarged gland with a round or ovoid shape, with high or mixed attenuation. Contrast enhancement is usually not seen. 10 Later, the adrenal hemorrhage may appear as an isoattenuating gland that often calcifies after 1 year. MRI can accurately determine the timing of the development of the hemorrhage; if the images are taken less than 7 days after hemorrhage (acute phase), the blood is hyperintense on T1-weighted images and hypointense on T2-weighted images. Between 7 days and 7 weeks (subacute phase), the hypointense signal on T2-weighted images gradually changes to a hyperintense signal. After 7 weeks (chronic hematoma), both T1- and T2-weighted images show a hypointense signal for blood products.11,12 In our case, radiological examinations (CT and MRI) of the adrenal glands were consistent with subacute hemorrhage.
Steroid treatment must be initiated promptly to reduce the risk of death. In addition, anticoagulant treatment should be given in the context of APS because it reduces the risk of thrombosis in the future. 13
The patient with adrenal crisis requires management with parenteral corticosteroids. Hydrocortisone 100 mg bolus is usually used, followed by 200 mg every 24 h, administered as a continuous infusion or as IV or intramuscular boluses every 5 h.13,14 It is also recommended to administer IV fluids early (1000 mL within the first hour), with crystalloid fluids. 15 IV 5% dextrose is usually administered when glucose levels are below 70 mg/dL. Once the adrenal crisis has resolved, the hydrocortisone dose should usually be reduced to the usual maintenance dose more than a 3-day period. Mineralocorticoid substitution with fludrocortisone at a mean dose of 100 μg/day (typically 50–200 μg/day) is also recommended. 16
Vitamin K antagonists (VKA) are the main anticoagulant treatment for thrombotic APS. Based on the results of the rivaroxaban in thrombotic APS (TRAPS) trial, 17 EULAR (European Alliance of Associations for Rheumatology) recommends against the use of direct oral anticoagulants (DOAC) in patients with triple-positive APS. 18 The International Society on Thrombosis and Hemostasis (ISTH) recommends that DOAC not be used in high-risk APS (triple positive, previous arterial thrombosis) and in patients with recurrent thrombotic events despite an INR value in the therapeutic range. 19 Based on these recommendations, the patient was treated with warfarin once control of the hemorrhagic event in the adrenal gland was confirmed and no active bleeding occurred in any other system.
The patient’s elevated thyroid stimulating hormone (TSH) levels with normal free thyroxine (FT4) were consistent with subclinical hypothyroidism. To avoid overlooking the diagnosis of hypothyroidism or precipitating adrenal crisis in patients with adrenal insufficiency, routine testing for both abnormalities may be warranted in this patient population. 20 Subsequently, studies were supplemented with antithyroid peroxidase (TPO) antibodies and were negative. The patient was treated with thyroxine after being started on hydrocortisone to prevent persistence of adrenal crisis.
Regarding disease prognosis, there is a retrospective study of patients with BAH secondary to APS between January 1990 and July 2010, who had a favorable long-term outcome. One patient died of cerebral hemorrhage 3 months after the onset of adrenal insufficiency. Repeated synacthen tests showed complete absence of response in 8 of the 10 patients assessed. Although adrenal dysfunction is generally irreversible, in rare cases adrenocortical function may recover at least partially. Therefore, measurement of morning cortisol during follow-up is indicated to identify these patients. 21
Conclusion
Adrenal crisis is a rare manifestation of APS. Adrenal hemorrhage is the main cause of adrenal crisis due to APS. Given the high mortality rate that occurs when this disease is not diagnosed or treated, a high clinical suspicion is required. The aim of this publication is to highlight the importance of considering this differential diagnosis, as its nonrecognition would have catastrophic consequences. In our case, adrenal involvement was the first manifestation of this syndrome and the key to its subsequent diagnosis.
Footnotes
Acknowledgements
The authors thank the endocrinology department of the San Ignacio University Hospital.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical approval
Our institution does not require ethical approval for reporting individual cases or case series.
Informed consent
Written informed consent was obtained from the patient(s) for their anonymized information to be published in this article.