
Abstract
Acute lymphoblastic leukemia is typically characterized by leukocytosis, resulting from the uncontrolled proliferation of malignant cells. However, we report an atypical case of acute lymphoblastic leukemia that presented with leukopenia and exhibited a protracted clinical course spanning 6 months. The patient, a 45-year-old female, initially presented to our hospital with recurrent fever and was found to have lymphoblasts in a hypoplastic bone marrow. Upon further investigation, the patient was diagnosed with B-cell lymphoblastic leukemia, not otherwise specified, based on cell surface antigen expression and genetic abnormalities. Notably, the patient demonstrated persistently low white blood cell and neutrophil counts, without evidence of increasing lymphoblast infiltration in the bone marrow during the ensuing 6-month period. Subsequent chemotherapy led to normalization of hematopoiesis and disappearance of lymphoblasts, resulting in complete remission of the disease.
Keywords
Introduction
Acute leukemia is characterized by leukocytosis and hyperplastic bone marrow and typically progresses rapidly over a period of weeks due to the presence of highly proliferative tumor cells. Patients with secondary acute myeloid leukemia (AML) following myelodysplastic syndrome, aplastic anemia (AA), and myelofibrosis (MF) often present with pancytopenia and hypoplastic bone marrow, which progress slowly over several months. 1 Previous retrospective studies have indicated that a small proportion of AML cases, approximately 4.8%, are hypocellular, defined as having bone marrow cellularity ⩽20%. 2 In contrast, acute lymphoblastic leukemia (ALL) with hypoplastic bone marrow is a rare occurrence. A study involving pediatric ALL patients reported that 12.2% and 0.2%–2% of patients presented with pancytopenia and hypoplastic bone marrow, respectively. 3 However, in adults, only a few cases of ALL with hypoplastic bone marrow have been documented,4,5 and their clinical course and prognosis remain unclear. Herein, we describe a rare case of ALL presenting with leukopenia and progressing slowly over a period of 6 months. Due to the atypical clinical presentation, establishing a definitive diagnosis and developing an appropriate treatment strategy posed significant challenges. Therefore, we present this case along with a comprehensive review of the existing literature.
Case report
The patient was a 45-year-old female with an unremarkable medical history. Six months prior to her presentation, her complete blood cell count (CBC) was within normal limits, with a white blood cell count of 5500/µL, hemoglobin level of 13.7 g/dL, and platelet count of 264 × 103/µL. The patient presented to a local hospital with a 1-month history of intermittent fever and was found to have leukopenia on CBC, with a white blood cell count of 1600/µL and a neutrophil count of 600/µL. The patient was subsequently referred to our hospital for a comprehensive evaluation.
During the patient’s initial visit, baseline demographics were obtained (height: 158 cm; weight: 44.5 kg; blood pressure: 109/77 mm Hg; heart rate: 101 beats/min; and temperature: 36.6°C). Upon auscultation, normal heart, and breath sounds were observed, and there was no evidence of abdominal tenderness, edema, or superficial lymphadenopathy.
The CBC revealed neutropenia and thrombocytopenia, without any evidence of anemia. Peripheral blood smears showed no blasts or other morphological abnormalities. Coagulation parameters were within normal limits, and biochemical testing indicated normal levels of lactate dehydrogenase, C-reactive protein, and soluble interleukin-2 receptor (Table 1). In addition, paroxysmal nocturnal hemoglobinuria (PNH)-type cells were detected in less than 0.003% of the granulocytes and erythrocytes. Bone marrow aspiration demonstrated slightly hypoplastic bone marrow (Figure 1(a)), with a nucleated cell count of 14,600/µL, including 37% small lymphoblasts (Figure 1(b)). Flow cytometry (FCM) revealed the following cell populations: CD2 (−), CD3 (−), CD7 (−), CD10 (+), CD13 (−), CD19 (+), CD20 (−), CD22 (+), CD33 (−), CD34 (−), CD38 (+), HLA-DR (+), MPO (−), and TdT (+), without expression of light chain. Bone marrow biopsy (Figure 1(c)–(j)) showed normocellular bone marrow with 50% cellularity, with reduced number of granulocytic cells and few megakaryocytes and erythroblastic islands observed. Immunostaining demonstrated that 30%–50% of the nucleated cells were blast-like cells with CD3 (−), CD10 (+), CD20 (−), CD79a (+), CD34 (±), and TdT (+), indicating the presence of precursor B-cell ALL. Furthermore, a silver staining technique showed mild increases in reticular fibers. Notably, no leukemia chimeric genes were detected, and the patient exhibited a normal karyotype. Through polymerase chain reaction (PCR), we detected monoclonal gene rearrangement of immunoglobulin heavy chain, which confirmed clonality of the lymphoblasts. In addition, we employed case-specific primers to evaluate bone marrow minimal residual disease (immunoglobulin-PCR). Computed tomography revealed no evidence of lymphadenopathy, and bone marrow magnetic resonance imaging (MRI) revealed a homogeneous bone marrow signal without fatty marrow.
Laboratory findings at the first visit to our hospital.

Hb: hemoglobin; WBC: white blood cell; Neu: neutrophil; Eo: eosinophil; Ba: basophil; Mono: monocyte; Lym: lymphocyte; PLT: platelet; PT-INR: prothrombin time-international normalized ratio; FDP: fibrin degradation product; T-Bil: total bilirubin; AST: aminotransferase; ALT: alanine aminotransferase; LDH: lactate dehydrogenase; CRE: creatine; CRP: C-reactive protein; M-protein: monoclonal protein; IgG: immunoglobulin G; IgA: immunoglobulin A; IgM: immunoglobulin M; sIL-2R: soluble interleukin-2 receptor; ANA: antinuclear antibody; EBV: Epstein-Barr virus; VCA: virus capsid antigen; EBNA: EBV nuclear antigen; CMV: cytomegalovirus; Parvo: human parvovirus B19.
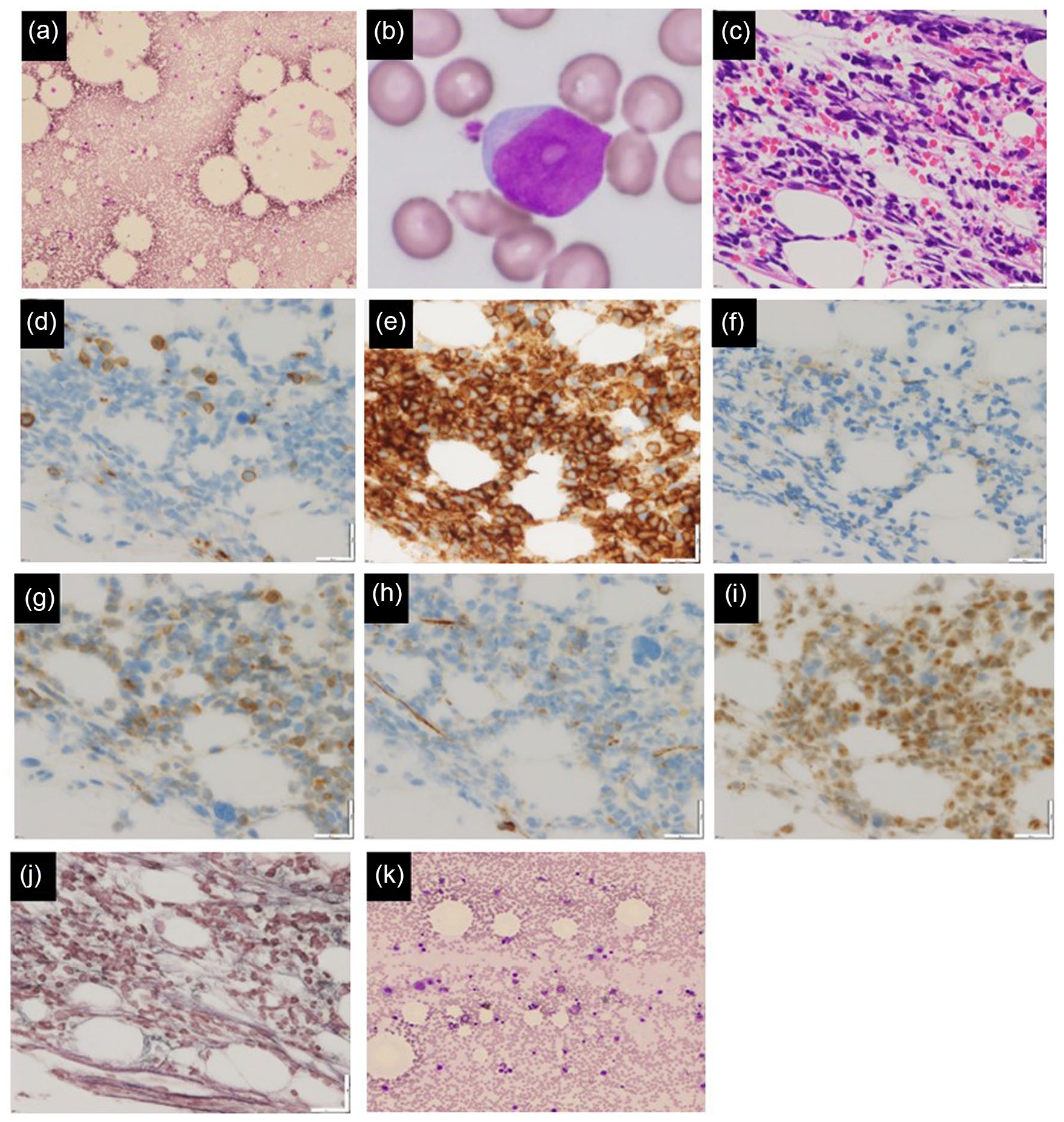
(a, b) Bone marrow smear during the initial examination, (c)–(j) Bone marrow biopsy, (k) Bone marrow smear on the complete remission. ((a) Smear (100×); hypoplastic bone marrow with lymphoblast. (b) Smear (1000×); small lymphoblast with agranular and distinct nucleoli. The chromatin is not condensed and the nucleocytoplasmic ratio is high. The cytoplasm is moderately basophilic. (c) Hematoxylin and eosin staining (40×); normocellular bone marrow with cellularity of 50%. Lymphoblast comprises 30%–50% of the nucleated cells. (d) CD3 (40×); negative. (e) CD10 (40×); positive. (f) CD20 (40×); negative. (g) CD79a (40×); positive. (h) CD34 (40×); weakly positive. (i) TdT (40×); positive. (j) Silver staining (40×); mild increase in reticular fibers. (k) Smear (200×); normocellular bone marrow without lymphoblast.)
A diagnosis of B-cell lymphoblastic leukemia (NOS) was established through the analysis of morphological and FCM findings, as well as gene abnormalities. However, the presence of low white blood cell count and slightly hypoplastic bone marrow were deemed atypical for ALL. Following the confirmation of an HLA-matched sibling donor, chemotherapy was initiated 6 months after the initial visit (Figure 2). The patient underwent induction therapy, which comprised of prednisolone (60 mg/m2, days 1–28), daunorubicin (45 mg/m2, days 8–10), cyclophosphamide (1200 mg/m2, day 8), vincristine (1.3 mg/m2, days 8, 15, 22, and 29), and
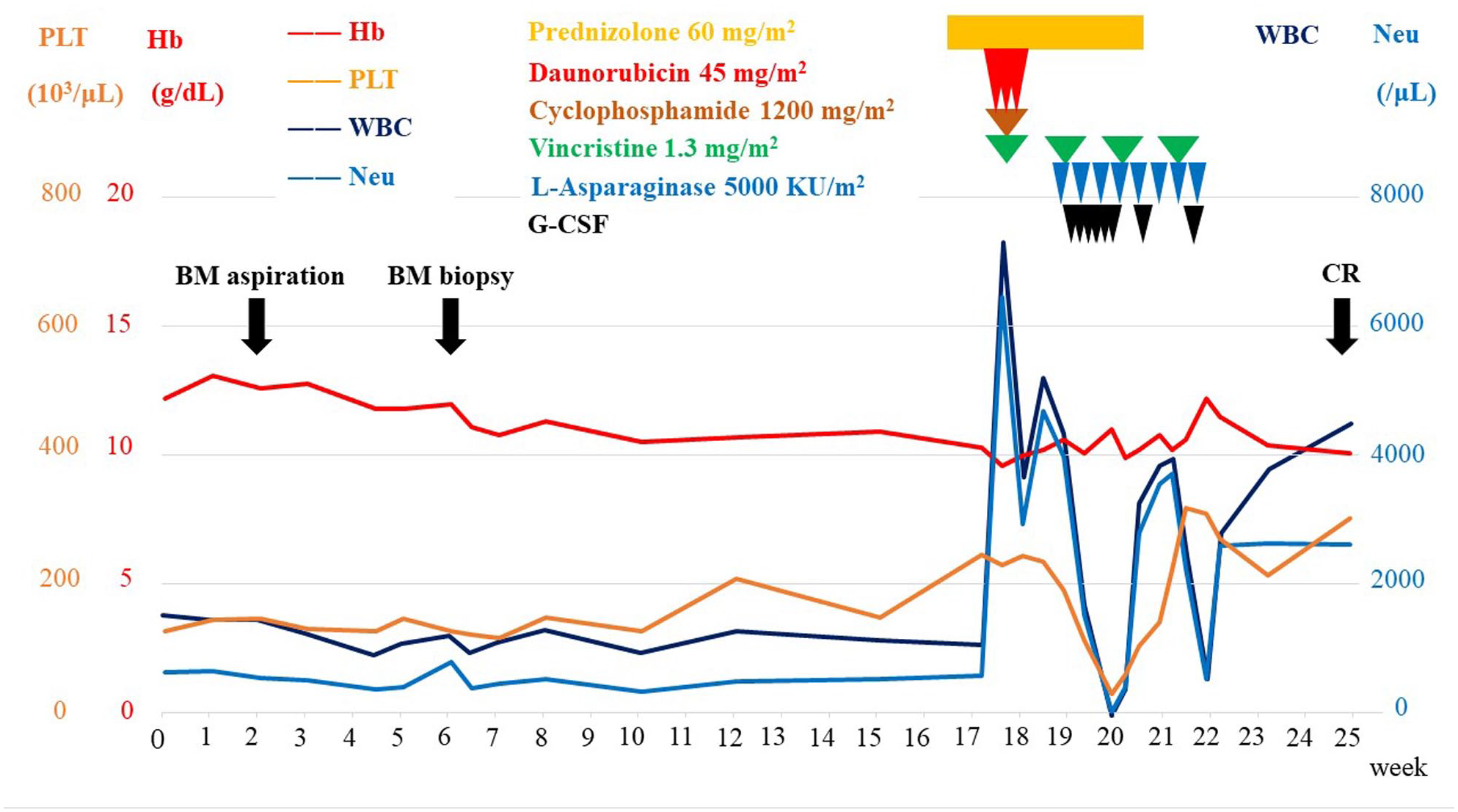
Clinical course from the first visit.
Discussion
This report details a unique case of ALL characterized by leukopenia and a slow progression spanning 6 months. Following chemotherapy, the disappearance of lymphoblasts disappeared and the restoration of normal hematopoiesis were observed.
Typically, ALL presents as leukocytosis and hyperplastic bone marrow, progressing rapidly within a matter of weeks. However, our patient exhibited an atypical presentation, as evidenced by the slightly hypoplastic bone marrow and the absence of an increase of lymphoblasts over the 6-month period. Consequently, we diagnosed the patient with relatively hypocellular ALL, a remarkably infrequent occurrence.
Distinguishing hypocellular ALL from ALL subsequent to AA or MF is essential. Notably, a prior investigation reported several instances of pediatric ALL preceded by bone marrow failure, 6 while a few cases of adult ALL were similarly preceded by bone marrow failure. 7 In this particular case, although the bone marrow exhibited mild fibrosis, we were unable to diagnose MF. Similarly, a diagnosis of AA was improbable, as evidenced by the absence of PNH-type blood cells and fatty marrow, as observed on MRI.
Morphological differentiation of hematogones and B-cell ALL presents a challenging task. 8 Hematogones, which are normal B-cell precursors, constitute a polyclonal population and are frequently observed during bone marrow recovery after chemotherapy, viral infection, and autoimmune cytopenia. Nonetheless, it has been reported that certain patterns of cell surface antigens may allow for the differentiation of hematogones. 9 In this study, however, we were unable to differentiate hematogones based on morphological and cell surface antigenic characteristics. Nevertheless, we established a diagnosis of B-cell ALL by detecting monoclonal genetic rearrangement of immunoglobulin heavy chain.
Following a 6-month period, induction chemotherapy was administered to verify the existence of a human leukocyte antigen-matched sibling donor due to concerns of potential delay in bone marrow recovery in the patient. In the context of pediatric ALL, a prior investigation revealed that there was no significant difference in overall survival outcomes between hypercellular and hypocellular ALL. 3 In contrast, a separate study indicated that adult hypocellular ALL demonstrated a gradual progression toward hypercellularity between 6 months to 1 year. 4 Despite this, there is currently no evidence to suggest that treating hypocellular ALL before transitioning to hypercellular ALL results in improved treatment outcomes. In this particular case, the patient was treated prior to progression due to the presence of intermittent fever and worsening of general health status.
Conclusion
In conclusion, we present a case of ALL characterized by a slow progression and associated leukopenia. While the establishment of a definitive diagnosis and corresponding treatment plan presented significant challenges, the patient ultimately demonstrated restored normal hematopoiesis following chemotherapy.
Footnotes
Acknowledgements
The author thanks the patient who agreed to present her case in this report.
Author contributions
M.S. and K.I. participated in patient treatment and collection of relevant data; S.T. and H.H. reviewed the literature and participated in the drafting of the manuscript; and all authors issued final approval for the version to be submitted.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics approval
Our institution does not require ethical approval for reporting individual cases or case series.
Informed consent
Written informed consent was obtained from the patient for her anonymized information to be published in this article.
Patient consent for publication
Written informed consent was obtained from the patient for the publication of this case report.