
Abstract
Castleman disease is a heterogeneous group of disorders of lymphoid tissue. It can often mimic other autoimmune diseases or malignancies, presenting a diagnostic dilemma to attending clinicians. Systemic lupus erythematosus and Castleman disease share many clinical and biochemical features rendering a special diagnostic challenge. Herein, we report a case of a young female who initially presented with features of idiopathic multicentric Castleman disease, subsequently developed antinuclear antibody positivity, and fulfilled the criteria for the diagnosis of systemic lupus erythematosus. Following the commencement of treatment for systemic lupus erythematosus, she had a marked response with resolution of altered clinical and biochemical profile.
Keywords
Introduction
Systemic lupus erythematosus (SLE) is a multisystem autoimmune disorder that predominantly affects young females. It might present with an array of clinical features ranging from constitutional symptoms to sinister neurological features such as seizures.
Castleman disease (CD), first described in the 1950s by Dr Benjamin Castleman, is a lymphoproliferative disorder, which encompasses heterogeneous clinical and pathological features that overlap between autoimmune, infectious, and malignant disorders. 1 The spectrum ranges from autoimmune diseases including SLE, rheumatoid arthritis, and juvenile idiopathic arthritis (JIA) to malignant disorders such as lymphoma, sarcoma, and POEMS syndrome (Polyneuropathy, Organomegaly, Endocrinopathy, Monoclonal gammopathy, and Skin changes). 2
Diagnosing either of these diseases (SLE and CD) in a patient with nonspecific symptoms carries a concomitant risk of diagnostic inaccuracy requiring a carefully planned diagnostic workup.
This case report discusses a young female presenting with clinical features of idiopathic multicentric Castleman disease (iMCD) who developed antinuclear antibody (ANA) positivity later during the course of the disease, subsequently diagnosed with SLE (with nephropathy). She responded well to SLE-targeted therapy.
Case report
A 25-year-old recently married female presented to a tertiary care hospital in Anuradhapura, Sri Lanka, with a 3-week history of fever, malaise, cough, shortness of breath, and loss of appetite with mild hair loss for a few months. Her past medical history was unremarkable although one of her monozygotic triplet sisters had been diagnosed with SLE. On examination, she was febrile, and pale, with multiple lymphadenopathies involving the cervical, axillary, and inguinal groups along with facial puffiness and bilateral lower limb edema. Bi-basal fine end-inspiratory crepitation with hepatosplenomegaly was also noted.
Investigations revealed a low hemoglobin level and thrombocytopenia, elevated erythrocyte sedimentation rate (ESR), C-reactive protein (CRP), and lactate dehydrogenase levels (LDH) (Table 1).
Initial investigation results.
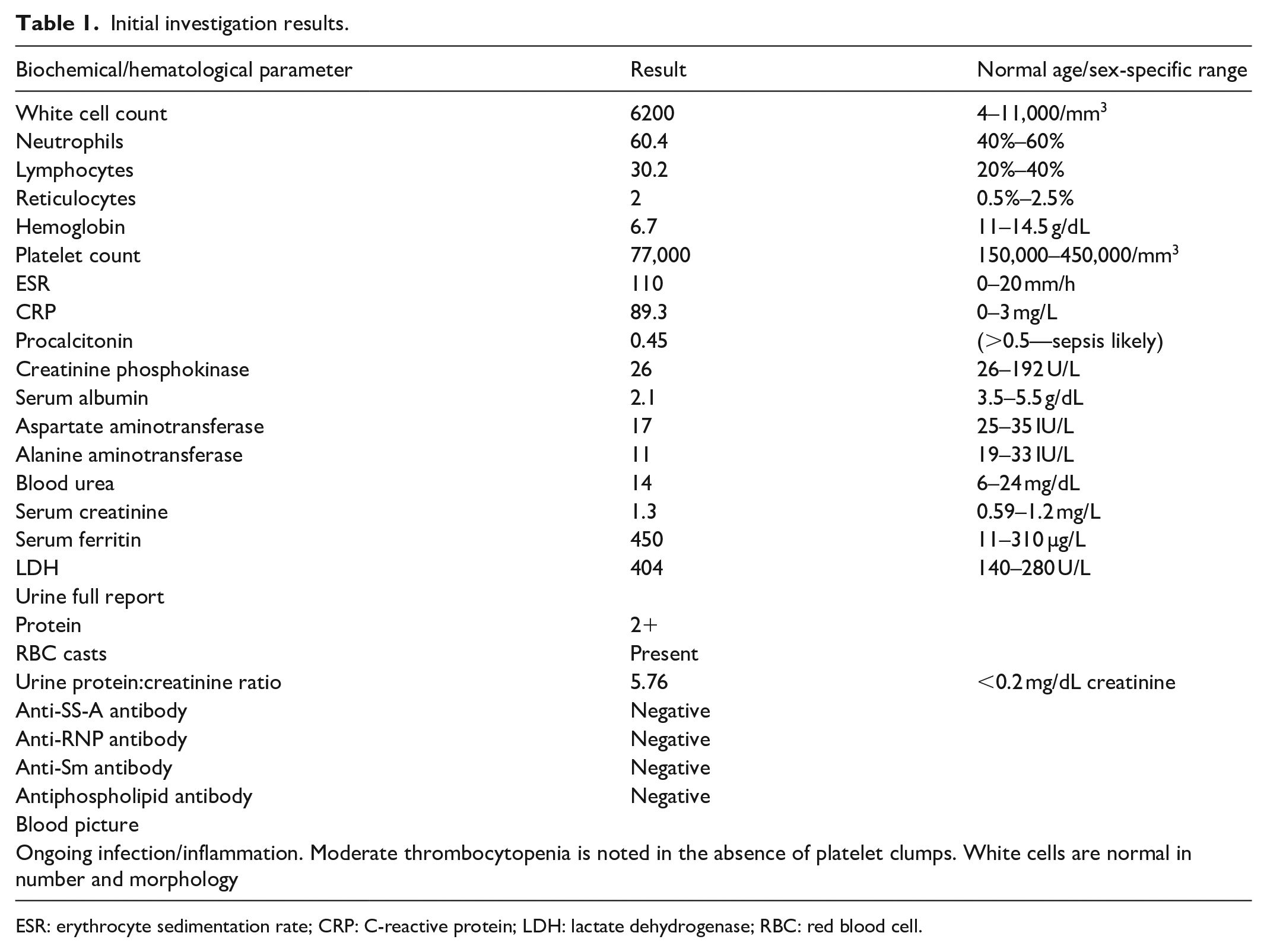
ESR: erythrocyte sedimentation rate; CRP: C-reactive protein; LDH: lactate dehydrogenase; RBC: red blood cell.
Urine analysis showed proteinuria with red blood cell (RBC) casts. The 24-h urine protein quantification confirmed nephrotic range proteinuria and the urine protein:creatinine ratio (U:PCR) was 5.76 mg/dL. Bilateral pleural effusions and a left lower zone consolidation were noted in the chest x-ray. Ultrasound abdomen confirmed hepatosplenomegaly. Blood, urine, and sputum cultures were negative. She was started on intravenous co-amoxiclav and clarithromycin and two units of leukodepleted blood were transfused slowly.
The initial ANA titer was negative. Conversely, the repeated test done 3 weeks later was found to be positive (titer—1:160), whereas simultaneous C3 (20 mg/dL) and C4 (10 mg/dL) complement levels were low (reference ranges: 50–120, 20–50 mg/dL, respectively).
Anti-double-stranded DNA, virological studies for SARS-CoV-2, human immunodeficiency virus, Epstein–Barr virus (EBV), cytomegalovirus, hepatitis B and C were all found to be negative. Sputum, Mantoux, and polymerase chain reaction testing for tuberculosis yielded negative results. There was no echocardiographic evidence of infective endocarditis.
Lymph node biopsy histology revealed reactive secondary follicles with hyalinization which were suggestive of CD. Bone marrow biopsy was negative for hematological malignancies or plasma cell disorders. Serum protein electrophoresis was negative for monoclonal gammopathy. The patient was referred to the consultant rheumatologist and was started on intravenous methylprednisolone pulse therapy (1 g per day for 3 days), oral steroid 45 mg per day, and hydroxychloroquine. She was taken over by the nephrology team for further management of renal complications of SLE. A dramatic improvement was noted clinically and biochemically with the normalization of inflammatory markers and platelet counts with the gradual but steady decline of proteinuria which has been normalized in 1-month review by the medical team (Graph 1(a) and (b)). Due to a satisfactory response to steroid therapy, renal biopsy was not opted for by the nephrology team at this point. Two months following her discharge, she was readmitted with pleurisy with pleural effusion suggestive of serositis. She was commenced on cyclophosphamide therapy as an adjunct by the pulmonologist.

Trends of investigation profile (red arrow depicts the methylprednisolone pulse therapy). (a) trends of inflammatory markers and (b) trends in main cell lines and urine proteinuria.
Discussion
Primarily, the CD has been divided into unicentric (with localized lymph node enlargement and minimal symptoms) or multicentric disease which involves heterogeneous lymph node groups and other lymphoid organs possessing a variable symptomatology.2,3 Exact etiopathogenesis of this diverse disease has not yet been established but viruses like HHV-8 have been postulated especially in MCD, whereas HHV-8-negative MCD is categorized as iMCD. 2 Diagnosis of CD is a clinical challenge largely due to the heterogeneity of clinical presentation. Moreover, a vast number of diseases could mimic this peculiar disease. 4
The conclusive diagnosis of CD is made following the exclusion of these conditions, in the presence of two major criteria (which are compulsory) in combination with 2 of 22 minor criteria. 2 The non-exhaustive list of conditions that mimic CD includes infectious disorders such as infectious mononucleosis or chronic active EBV, inflammation, and lymphadenopathy caused by other uncontrolled infections; autoimmune diseases (SLE, rheumatoid arthritis, adult-onset Still’s disease, JIA); lymphoproliferative disorders such as lymphoma (Hodgkin and non-Hodgkin) or plasma cell disorders including multiple myeloma; and POEMS syndrome. 2 SLE (which should be diagnosed based on full criteria rather than autoantibodies alone) is one of the diseases categorized under exclusion criteria before diagnosing CD.
Three main histological lymph node patterns in CD have been identified so far, which include hyaline vascular and plasma cell infiltration pattern, representing two ends of a spectrum of histological variance rather than two distinct entities, along with a the mixed pattern which represents the center of the spectrum. 2
According to the most recent EULAR/ACR (European League Against Rheumatism/American College of Rheumatology) classification criteria (2019), SLE diagnosis can be made in the presence of positive ANA titer along with other clinical, biochemical, and renal histological findings. 5 Although lymphadenopathy could be observed in SLE, it does not constitute a diagnostic criterion, eliminating the need to examine the lymph nodes for diagnostic purposes. However, studies have shown multiple histological features in lymph nodes in SLE which include lymph node necrosis, reactive follicular hyperplasia with or without giant follicles, nonspecific follicular hyperplasia, and atypical lymphoplasmacytic immunoblastic proliferation. 6 Interestingly, a minority of patients have shown histological features similar to that of CD as well.6,7 This fact validates the need to exclude SLE in patients who are suspected to have CD even when the major criteria for CD are already met.
In the case of our patient, she satisfied the diagnostic criteria for CD at presentation. As the disease evolved, she developed ANA positivity with low complement levels and fulfilled the diagnostic criteria for SLE also. Along with a strong family history of SLE (in her triplet kindred), and clinical and biochemical evidence fulfilling diagnostic criteria with the patient’s dramatic response to treatment, it was concluded this as a case of SLE with nephropathy.
It is noteworthy that the distinction between these two entities (SLE vs CD) can be challenging as the sensitivity of ANA can be as low as 70%, especially at the early stages of SLE, creating a time window where clinicians might disregard SLE as the diagnosis. 8 On the other hand, when the typical sero markers of SLE are absent and the prevalence of ANA in CD is not very well understood, concluding a diagnosis based on clinical features which are essentially shared by both diseases could lead to erroneous clinical judgment. There had been several case reports in the literature where CD is co-existent with other autoimmune diseases.9–12 To arrive at the diagnosis of CD by exclusion, the use of diagnostic criteria might pose a diagnostic dilemma. Changing diagnostic criteria over time also adds to this dilemma. Such diagnostic labeling can be different from the updated criteria, especially for a patient with a shifting clinical or biochemical profile, and might add to the confusion as retrospective data are also utilized for the diagnosis. In such settings, surrogate evidence such as family history and response to treatment can be of use.
The characteristic feature in this case report is the initial negative ANA titer leading to the initial exclusion of SLE which led to the diagnosis of CD. Due to strong positives toward SLE and nephrotic range proteinuria suggestive of nephropathy, repeated ANA was carried out to decisively rule out SLE as renal involvement is uncommon in CD. 13 With a brief note on the management, iMCD may need strong immunosuppressive therapy (biological agents, for instance) while SLE often responds to steroids and other traditional immunosuppressants.
Conclusion
SLE and CD are two distinct disorders that share many common clinical and biochemical features. However, the correct diagnosis and distinction between the two are crucial as management (which depend on the severity of each disease) differs significantly. The need for further research and development of more disease-specific, efficient serological markers which will enable the early diagnosis of CD to save precious time in managing critically ill patients who need urgent medical interventions cannot be overemphasized.
Footnotes
Acknowledgements
The authors would like to acknowledge the Department of Pathology and Department of Nephrology of our institution for their contributions during the management of this patient.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical approval
Our institution does not require ethical approval for reporting individual cases or case series.
Informed consent
Written informed consent was obtained from the patient(s) for their anonymized information to be published in this article.