
Abstract
Membranous glomerulonephritis is the most common glomerular disease in adults. Its primary form has been characterized with formation of phospholipase A2 receptor antibodies. Malignancy, infections, and autoimmune disorders are the most common causes of secondary membranous glomerulonephritis. We present a case of a 55-year-old African American female who presented with nephrotic range proteinuria and diagnosed with secondary membranous glomerulonephritis based on distinct pathological features on kidney biopsy and absence of serum phospholipase A2 receptor antibodies. She initially underwent extensive workup for malignancies, infections, and common autoimmune disorders which were all negative. Her proteinuria remained resistant to steroid treatment and she was treated with subcutaneous adrenocorticotropic hormone injections. Meanwhile, she was also diagnosed with the anti-muscle specific kinase antibody variant of myasthenia gravis. In literature, there are few case reports of myasthenia gravis as a cause of secondary membranous glomerulonephritis. In our case, the lack of other inciting factors also suggested this association.
Keywords
Introduction
Membranous glomerulonephritis (MGN) can be a primary condition or secondary to various exposures with each type having distinct pathological features. Secondary MGN can be associated with malignancy as well as other autoimmune, infectious, and drug-related exposures (such as non-steroidal anti-inflammatory drugs (NSAIDs) and interferon).1–4 Clinically, both primary and secondary MGN are typified by high-grade proteinuria and a nephrotic presentation though hematuria may rarely be seen along with severe proteinuria. Severe hypoalbuminemia and increased risk of venous thromboembolism are hallmarks of MGN due to loss of small proteins and peptides that regulate the clotting cascade. 5
Primary MGN is associated with anti-phospholipase A2 receptor (PLA2R) antibodies and IgG4 antibody deposition, while secondary disease presents mesangial deposits and IgG1 subtype of immunoglobulins (Ig) in malignancy and other Ig subtypes in non-malignant secondary MGN. 6 Thyroiditis, colitis, syphilis infection, and many types of drug exposures as stated above can be associated with development of secondary MGN. Pathologically, the disease appears similar with spike and ball pattern of antibody deposition along epithelial cells on electron microscopy, granular IgG immunofluorescence, and thickened glomerular basement membrane (GBM) on light microscopy.1–4 Anti-PLA2R can usually be detected on renal biopsy and serologically in 70% of primary MGN cases, and anti-thrombospondin domain type 1–containing 7a (Anti-THSD7A) is present in 10% of cases without anti-PLA2R. Primary MGN cases do not usually include mesangial deposits, which is a feature of secondary presentations of MGN. 7
Myasthenia gravis (MG) is a chronic autoimmune neuromuscular disease that can be associated with various immunological disorders. Recent evidence supports the association between nephropathy and thymic diseases (thymoma, thymic hyperplasia, and MG). Minimal change disease (MCD) has been reported most often with a few reported cases of secondary MGN seen in association with MG. 8 Here, we report a rare case of combination of MG with secondary membranous nephropathy.
Case report
Our case is 56-year-old female with a past medical history significant for squamous cell carcinoma of the cervix for which she had partial hysterectomy in 2000, and vaginal cancer in 2006 treated with radiation, presented with a new-onset high-grade proteinuria (13 g/day on 24-h urine) and up to 11.8 g urine protein/g creatinine on urine protein to creatinine ratio. She also clinically manifested with anasarca, hypoalbuminemia (albumin: 2.1 g/dL) and hyperlipidemia (low-density lipoprotein (LDL): 379 mg/dL) since September 2016, all consistent with nephrotic syndrome.
Clinically, the patient reported diplopia, weakness, and a decrease in exercise tolerance about at about the time of presentation (September 2016). She had been an avid marathon runner prior to presentation in 2016, and now she reported hardly being able to run a mile or walk briskly. She had intact renal function at the time of the presentation with serum creatinine of 70.72 µmol/L (0.8 mg/dL) and blood urea nitrogen (BUN) of 4.64 µmol/L (13 mg/dL) but significant proteinuria warranted a renal biopsy while initial serology workup was pending. Patient had renal biopsy on October 2016 with a pathology report that showed secondary membranous nephropathy. The specimen was sent for PLA2R immunofluorescence testing. Initial serology workup showed positive a low-titer positive ANA (antinuclear antibody) of 1:80, a low-titer positive anti-dsDNA at 1:282 IU(international units)/mL, with normal 171 mg/dL C3 and 36 mg/dL C4, high erythrocyte sedimentation rate (ESR) (121 mm/h), normal thyroid-stimulating hormone (TSH), negative HIV abs and negative Hepatitis B surface Abs and Antigens, and negative rheumatoid factor (RF). Serological testing for Anti-PLA2R and Anti-THSD7A both returned as negative as well.
The patient was started on Furosemide 40 mg twice daily, Pravastatin, Ergocalciferol 50,000 units orally weekly (for low vitamin D level), Losartan 25 mg daily, Aspirin 325 mg (after she has done the renal biopsy) because of the risk of hypercoagulation, and was instructed to be on low salt diet.
The patient obtained a PET-CT (positron emission tomography–computed tomography) scan from the skull base to the mid-thighs that revealed enlarged left axillary lymph node (24 × 16 mm) that is amenable to US-guided percutaneous biopsy, enlarged left subpectoral lymph node, multiple retroperitoneal/left para-aortic nodes, left common iliac lymph nodes/left internal iliac lymph nodes. The lymph node biopsy was negative for lymphoma and only showed the rest of the serology workup such as anti-neutrophil cytoplasmic antibody (ANCA), anti-phospholipid antibody (APLS), anti-cardiolipin (anti-CL) antibody, scleroderma antibodies (SSA/SSB), and anti-GBM antibodies were all negative.
The immunofluorescence staining of the renal biopsy specimen for PLA2R revealed no glomeruli are present on the section stained for PLA2R (inconclusive result); however, the presence of mesangial deposits in the biopsy suggested secondary MGN which was due to relapsing malignancy or an associated autoimmune condition. The immunofluorescence showed IgG and C3, but did not show the “full house” pattern of C3, C1q, IgM, IgG, IgA, excluding a diagnosis of membranous lupus nephritis. See Figure 1 for summary of slides and findings suggesting secondary MGN.
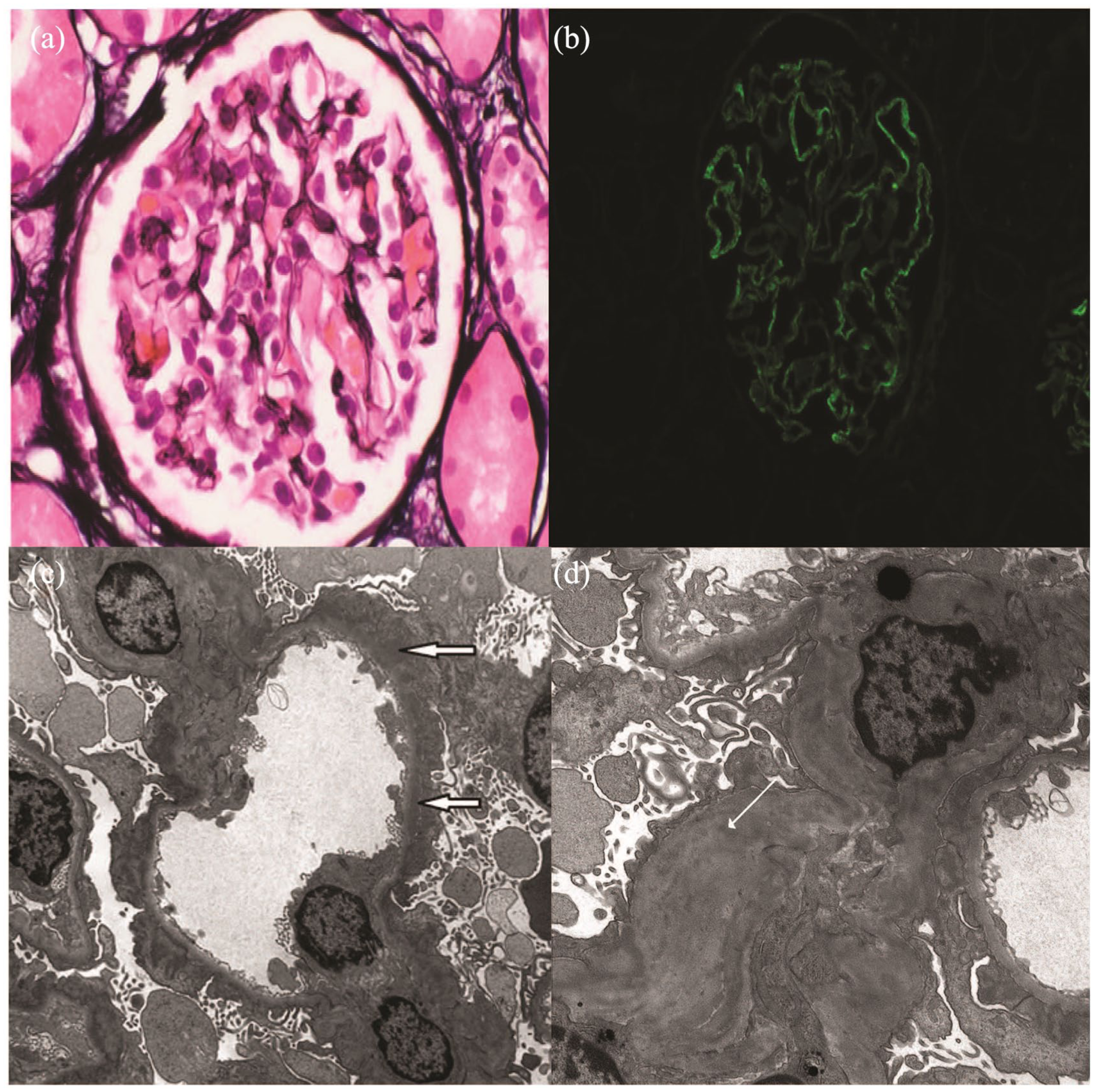
Renal biopsy slides showing secondary glomerulonephritis: (a) light microscopy showed glomerulus with normal size and cellularity. Basement membrane spikes are not observed, consistent with early stage membranous nephropathy (Jones methenamine silver stain; original magnification 600×). (b) Immunofluorescence showed granular capillary wall staining for IgG at 2+ intensity (IgG direct immunofluorescence stain; original magnification 400×). (c) Electron microscopy demonstrated glomerular capillary with numerous small subepithelial electron-dense immune complex deposits (arrows) adjacent to occasional, very small, spikes of basement membrane material (Electron microscopy study; original magnification 5000×). (d) Electron microscopy demonstrating mesangial deposits (white arrow) (Electron microscopy study; original magnification 5000×).
Since symptoms and serologies for systemic lupus were negative, and given the lymphadenopathy on PET-CT scan, concerns of relapsing malignancy were raised. Cancer surveillance testing was needed which included biopsy of the FDG-avid L.N, mammography, colonoscopy and bone marrow aspiration.
On 9 January 2017, CT-guided biopsy was done; bone marrow aspiration was done on 24 March 2017; and beside mammography and colonoscopy, all revealed no signs of malignant relapse. Meanwhile, the dose of Losartan was increased and she was switched from furosemide to bumetanide as the latter had longer half-life.
Since May 2017, the patient started to feel more and more fatigued with complaints of weak nasal voice, and on June 2017, the patient started to note episodes of diplopia. According to the patient, she had the first episode of vertical diplopia in 2000 that lasted for few weeks and disappeared with few flares over the last 17 years but the most recent one was on June 2017.
She was referred to a neurologist for full neurological evaluation and was started on prednisone 60 mg with plans to switch to ACTHAR® (repository corticotropin injection). The steroid was tapered off, and on August 2017, the patient started ACTHAR® with plans of 6–12 months treatment course with slow titration once proteinuria is controlled. On September 2017, the neurological assessment revealed MG with negative anti-acetylcholinesterase receptor antibodies but positive anti-muscle specific kinase (anti-MuSK) antibody and positive repetitive nerve stimulation (RNS) testing. While a RNS test was reported as positive in charting, the tracings were not immediately available to us. Anti-titin antibody was not specifically tested in this workup, but anti-LDL receptor-related protein 4 (LRP4) is negative. JC virus testing to look for an alternate diagnosis, like progressive multifocal leukoencephalopathy (PML), importantly, was negative. MG grading clinically fluctuated between class IIa and IIb by the Myasthenia Gravis Foundation of America (MGFA) classification.
She had no bulbar symptoms, no trouble with speech or respiratory distress, and no extremity weakness in the past either. ACTHAR® was tapered off gradually with a switch to rituximab for anti-MuSK MG. B cell depletion has resulted in marked remission of her neurological symptoms due to MG as well as of her proteinuria.
With this regimen of treatment, the patient has showed a remarkable remission clinically and with the level of proteinuria (from 13 g/day at the time of presentation to 0.59 g/day). While albumin rose to 2.9 g/L on adrenocorticotropic hormone (ACTH), after rituximab, it nearly normalized to 3.7 g/L. After stopping ACTH, urine protein to creatinine level increased to 5.4 g protein/g creatinine (UPC) only to enter complete remission after treatment with rituximab. See Figure 2 for graph of urine protein to creatinine ratio and onset of symptoms, start and stop of corticosteroid treatment, trend of proteinuria in response to ACTH treatment, and start date of rituximab (4 April 2018).

Graph of urine protein to creatinine ratio trend (g protein/g creatinine): black arrow shows start of symptoms of weakness and diplopia in September 2016 near initial diagnosis of nephrotic syndrome. Red arrow and line represent the initiation and duration of corticosteroid therapy (high dose 1 g/kg), and therapy end is denoted by end of red line. Green arrow and line indicate the start of adrenocorticotropic hormone therapy (ACTHAR©), 80 units twice a week dose, and therapy end is denoted by end of green line. Blue arrow and line indicate start of 0.375 g/m2 of body surface area (BSA) rituximab infusions weekly for a total of four infusions.
Discussion
MGN is a common cause of nephrotic syndrome in non-diabetic adults. 9 It usually occurs as idiopathic disease (primary MGN) due to kidney-limited autoimmune disorder. 10 It is characterized by GBM thickening without prominent hypercellularity, and the presence of subepithelial electron-dense deposits. 11
A large percentage of membranous nephropathy cases are, however, secondary to systemic autoimmune diseases such as systemic lupus erythematosus (SLE), infections, and the use of certain drugs (NSAIDs). 10 The mechanism by which the subepithelial deposits form in secondary MGN is not very clear, and can be explained by the deposition of circulating immune complexes in the subepithelial space. 10
MG is rarely associated with nephropathy. 12 In some cases of nephrotic syndrome, the mechanism is an imbalance between T helper cells class 1 and 2. 13 In MG, T cell dysfunction leads to the production of lymphokine, which increases the permeability of GBM. 14 Evidence supporting the association between nephropathy and thymic disease (thymoma, thymic hyperplasia, and MG) is growing, in which MCD is being the most common among other nephropathies.15–25
Clinically, distinguishing between primary and secondary MGN is of absolute importance, since therapy in the secondary MGN must be directed at the underlying cause, and some of the treatments for idiopathic MGN, like calcineurins (CNIs), are potentially toxic to the kidney. 9
Differentiating idiopathic MGN from secondary MGN, especially in elderly patients in whom malignancies tend to occur, necessitates the need for an accurate biomarker to use for differentiation.15,17 Studies have showed that the M-type PLA2R, a 185-kDa type I transmembrane glycoprotein expressed on glomerular podocytes, is the main target of autoantibodies in idiopathic MGN. 16 Subepithelial deposits form as antibodies bind to a membrane-based antigen and complement.9,10
The finding of mesangial deposits in our case raised the possibility of a secondary form of MGN. The specimen obtained from the renal biopsy was sent for PLA2R immunofluorescence testing to differentiate primary from secondary MGN which was non-conclusive, but subsequent serological testing for serum-anti-PLA2R was negative, supporting the diagnosis of a secondary membranous nephropathy. It is not impossible that the patient had an atypical form of primary MGN with a non-PLA2R IgG4 type antibody, but the findings of mesangial deposits in primary glomerulonephritis are rare, and the concomitant diagnosis of an autoimmune anti-MuSK MG is highly unusual. A unifying diagnosis of secondary MGN with appropriate mesangial deposition associated with a clinically concomitant diagnosis of anti-MuSK MG is more likely and more elegant.
Since the patient had no systemic manifestations of active SLE like joint pain, rash, oral ulcers and fever and the repeat ANA and anti-dsDNA were negative, the probability of lupus nephritis grade V was eliminated. Given her lymphadenopathy on PET-CT scan, concerns of relapsing malignancy were raised and cancer surveillance testing was needed which included biopsy of the FDG-avid L.N, mammography, colonoscopy and bone marrow aspiration.
In our case, the patient was started on prednisone and then switched to ACTHAR® (with a plan of 6–12 months of treatment) since steroids alone are not affective and ACTHAR® has been studied and was considered a safer alternative to CNI inhibitors and alkylating agents with reasonable efficacy in the treatment of membranous nephropathy.
Longer treatment course of ACTHAR® is hoped to induce remission with less risk than CNIs and steroids, rituximab, and certainly alkylating agents, unless renal function deteriorates acutely or proteinuria worsens steadily, then rituximab would be considered. The benefit of therapy using steroids and immunosuppressants suggests that membranous nephropathy is related to the disordered immune system. 12 Thymectomy, which is a standard therapy for MG, is still considered as an option for treatment. Thymectomy can be followed by numerous changes in lymphocyte functions which may require several years to be revealed, 24 suggesting that careful long-term follow-up for is necessary. Secondary membranous nephropathy can be, rarely, associated with thymic disorders. Therefore, clinicians should keep in mind that association; however, in our case, the symptoms of nephropathy have preceded the diagnosis of MG.
See Table 1 for previously reported cases of MG with or without thymic disease and glomerular disease.1,8,18,19,22,23,26–39 A newer case report by Bolz et al. 40 was added where the MGN could be classified as primary in setting of anti-PLA2R antibody positivity. There are 11 other cases with similar presentation to our currently presented case (total of 12 with current case): nine cases with MG and concomitant MGN and three cases with thymus disease (benign or malignant), MGN, but no active MG. A variety of other nephropathies that present with MG and both benign and malignant thymomas are also listed and totaled 70 cases.
Reported cases of thymic disease with or without myasthenia gravis and glomerular disease.

Avg: average; BT: benign thymoma; CC: current case; F: female; ECPGN: extra-capillary proliferative glomerulonephritis; FPGN: focal proliferative glomerulonephritis; FSGS: focal segmental glomerulosclerosis; g: gram; HSP: Henoch–Schonlein Purpura; IgA Neph: IgA nephropathy; LGN: lupus glomerulonephritis; M: male; MCD: minimal change disease; MG: myasthenia gravis; MGN: membranous glomerulonephritis; MPGN: membrano-proliferative glomerulonephritis, MT: malignant thymoma; N: no; n#: number of patients; neph: nephrotic range proteinuria (no amount specified); PGN: proliferative glomerulonephritis; Ref: reference; Y: yes.
Conclusions
We report a rare coexistence of autoimmune anti-MuSK MG and secondary MGN as defined by negative serologies and mesangial deposits on biopsy.
There have been cases reported with a primary MGN and MG as defined by the presence of anti-PLA2R and -THSD7 antibodies as well. 40
Treatment of anti MuSK MG resulted in concomitant resolution of proteinuria.
Secondary MGN can be due to autoimmune disease, not just malignancy, and treating concomitant autoimmune disease can result in a complete remission of proteinuria.
Footnotes
Authors’ Note
Authors confirm that this work contains no human research material. Patient agreed to publication of this report and provided documented written consent under condition of anonymity, with no publication of identifiable patient information. Ramy M Hanna is also affiliated with Division of Nephrology, Department of Medicine, UCI school of Medicine, Irvine, California.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical approval
Our institution does not require ethical approval for reporting individual cases or case series.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Informed consent
Written informed consent was obtained from the patient(s) for their anonymized information to be published in this article.