
Abstract
Objective:
The aim of this study is to determine the safety and the efficacy of amifampridine phosphate in muscle-specific kinase antibody-positive myasthenia gravis, in a 1:1 randomized, double-blind, placebo-controlled, switchback, double crossover study.
Methods:
Eligible patients had muscle-specific kinase myasthenia gravis, >18 years of age, and Myasthenia Gravis Foundation of America class II–IV with a score of ⩾9 on Myasthenia Gravis Composite scale. After the run-in phase, during which amifampridine phosphate was titrated to a tolerable and effective dosage, patients were randomized to receive placebo–amifampridine–placebo sequence or amifampridine–placebo–amifampridine sequence daily for 7 days. Then, patients switched treatment arms twice, for a total of 21 days of double-blind treatment. Safety was determined by serial assessments of adverse events/serious adverse events, physical examinations, and clinical and laboratory tests. The co-primary outcome measures included changes from baseline of Quantitative Myasthenia Gravis score and Myasthenia Gravis–specific Activities of Daily Living Profile score. The secondary outcome measures comprised changes from baseline of Myasthenia Gravis Composite score, Myasthenia Gravis Quality of Life scale—15 questions, Fatigue Severity Scale, and Carlo Besta Neurological Institute–Myasthenia Gravis scale. Statistical analyses were assessed using a switchback model for three-period, two-treatment crossover design.
Results:
A total of 10 patients were screened, enrolled, and treated. Transient paresthesias (60%) were the only amifampridine phosphate–related adverse events reported. Four patients were randomized to receive placebo–amifampridine–placebo sequence and three patients to receive amifampridine–placebo–amifampridine sequence. The co-primary objectives were statistically met (Quantitative Myasthenia Gravis score: p = 0.0003 and Myasthenia Gravis–specific Activities of Daily Living Profile score: p = 0.0006), as well as all the secondary endpoints (Myasthenia Gravis Composite score: p < 0.0001, Myasthenia Gravis Quality of Life scale—15 questions: p = 0.0025, Fatigue Severity Scale: p = 0.0061, and Carlo Besta Neurological Institute–Myasthenia Gravis scale: p = 0.0014).
Conclusion:
Despite the low number of patients, MuSK-001 study provided evidence that amifampridine phosphate, in the range of 30–60 mg daily dose, was safe and effective in treating muscle-specific kinase myasthenia gravis, suggesting the need for a large multi-center trial to confirm these results.
Introduction
Acquired myasthenia gravis (MG) is a rare auto-immune disorder of the neuromuscular junction (NMJ) characterized by fluctuating weakness and fatigability of skeletal muscles. MG pathogenesis relies on the binding of auto-antibodies to postsynaptic membrane proteins at the NMJ, which ultimately results in failure of neuromuscular transmission. Most commonly, MG is caused by complement-activating immunoglobulin (Ig) G1 and IgG3 to the acetylcholine receptor (AChR), with a consequent loss of functional AChR from the postsynaptic membrane. About 5%–10% of MG patients carry IgG4 anti-muscle-specific receptor tyrosine kinase (MuSK) antibodies.1–3
MuSK is located in the NMJ at postsynaptic level where it forms a complex with its co-receptor, low-density lipoprotein receptor-related protein 4 (LRP4). MuSK antibodies block the establishment of the agrin-LRP4-MuSK complex, which leads to disassembly of the AChR clusters, and altered NMJ maintenance and function. MuSK antibodies also interfere with the presynaptic compensatory mechanisms regulating acetylcholine (ACh) quantal release.2–5 The failure of this compensation might explain why MuSK-MG patients often show more severe forms of the disease, compared to AChR-MG patients. The different pathogenic mechanisms underlying MG produce wide variation in the presentation and severity of disease symptoms among MG patients. MuSK-MG is characterized by specific clinical features, such as female predominance, prominent oculo-bulbar involvement, and significant resistance to therapies.1,6
Conventional long-term treatments for MuSK-MG patients include acetylcholinesterase inhibitors (anti-AChE) and immunosuppressants, which often produce an insufficient response. The use of anti-AChE can exacerbate the NMJ impairment induced by the anti-MuSK antibodies and is frequently detrimental in these patients.7–9 Hence, the search for additional therapeutic options to treat MuSK-MG is a medical need.
3, 4-Diaminopyridine (3,4-DAP) is a non-specific voltage-dependent potassium channel (Kv1.5) blocker; it causes depolarization of the presynaptic membrane at the NMJ and delays nerve repolarization. This results in a prolonged action potential, which enables an increased release of vesicles containing ACh into the synaptic cleft.9–11 The greater availability of ACh enhances neuromuscular transmission, providing improved muscle function.
A considerable amount of clinical experience with the phosphate salt of 3,4-DAP (amifampridine phosphate (AP), Firdapse®) has been gained, providing evidence for its efficacy and safety in different autoimmune and genetic NMJ diseases, including Lambert–Eaton myasthenic syndrome (LEMS) and congenital myasthenic syndrome (CMS).12,13 Generally, AP has been recommended as first-line symptomatic treatment for LEMS by the European Federation of Neurological Societies (EFNS). 14
Based on MuSK-MG-specific pathogenic features, 2 recent nonclinical studies examined the effects of 3,4 DAP in MuSK mouse models, demonstrating that 3,4 DAP significantly improved neuromuscular transmission.9,15
Furthermore, anecdotal evidence suggested that AP was beneficial in a MuSK-MG patient 16 and in two AChR-MG patients, 17 but no randomized clinical trial (RCT) has been performed to date.
Here, we report the first phase-II RCT (MuSK-001) to determine safety, tolerability, and clinical efficacy of AP in the treatment of patients with MuSK-MG.
Methods
MuSK-001 was a randomized, double-blind, placebo-controlled, monocentric, crossover, outpatient, phase IIb clinical trial of AP in MuSK-MG. This study was conducted in Italy at the Istituto Neurologico Carlo Besta, in accordance with the ethical principles established by the Declaration of Helsinki, and the ICH Harmonized Tripartite Guideline: Guideline for Good Clinical Practice E6 (ICH E6). The study was approved by the Ethical Committee of the Istituto Neurologico Carlo Besta, and written informed consent was obtained from each patient screened for eligibility. This trial was registered at the Italian Agency for Drug (AIFA) Observatory with the number 46/2015. Trial protocol is available in EU Clinical Trials Register (EudraCT number: 2015-003127-62).
Patients
Patients included in the study were ⩾18 years of age, with a definitive diagnosis of MG-positive for the anti-MuSK antibodies serologic test. Eligible patients fell into class II–IV of Myasthenia Gravis Foundation of America (MGFA) Clinical Classification at Screening with a Myasthenia Gravis Composite (MGC) score equal or greater than 9 points at screening. All patients were required to have a stable steroid regimen for the month prior to screening and a stable ongoing immunosuppressive (e.g. azathioprine, mycophenolate mofetil, methotrexate, and cyclophosphamide) regimen for 3 months prior to screening. Ongoing treatments are reported in the “Results” section.
Exclusion criteria were as follows: weakness only affecting ocular or periocular muscles (MGFA class I) or intubation (MGFA class V); hypersensitivity to the active substance or to any of the excipients; epilepsy, uncontrolled asthma, or long QT syndrome; concomitant use of sultopride or any drug known to cause QTc prolongation; history of thymectomy within 12 months before screening; treatment with plasma exchange (PE) and/or intravenous immunoglobulin G (IVIG) within 3 weeks before screening, use of rituximab within 6 months before screening, employment of any investigational drug, device, or biological agent within 30 days before screening or while participating in this study.
Interventions
During the run-in phase of the study, the dose of AP (Firdapse; Catalyst Pharmaceuticals, Inc., Coral Gables, FL, USA) was determined for each patient by the investigator, within the bounds of a total daily dose of 30–100 mg, divided into doses taken 3–4 times per day, based on optimal neuromuscular benefit. The maximum single dose was 25 mg.
The identified dose and dosing regimen of AP remained stable for each patient throughout the randomization phase of the study.
Patients were randomly assigned in a 1:1 ratio to AP tablets or to placebo; both were administered orally, daily for 1 week at a time, according to the treatment arm. Patients in both groups were permitted to receive concomitant steroids and immunosuppressant therapies at their stable dose, as specified in the inclusion criteria. Treatment compliance was monitored by tablet accountability.
Study design and plan
As MuSK-MG is a rare disease, a crossover design, which enables the recruitment of a reduced number of patients, was adopted.
The design employed a two-treatment sequence: amifampridine–placebo–amifampridine (APA) and placebo–amifampridine–placebo (PAP) (Figure 1). The overall trial was at least 49 days, excluding the screening period which could last up to 14 days.

Schematic diagram of the MuSK-001 protocol.
Patients who successfully completed screening entered the MuSK-001 trial which included the following phases (Figure 1):
Open-label run-in. AP dose was titrated upward every 4 days, starting at 30 mg/day, at the discretion of the investigator, to identify the effective dose for each patient. Patient visits were scheduled at Day 1 of run-in phase, and every 2 weeks (±2 days) for a minimum of 3 weeks, with the last week requiring a stable dose and frequency. At the end of this period, patients must have had a 3-point improvement in MGC (excluding ocular items) 15 from Day 1 of run-in, to be eligible for randomization (Day 0).
Treatment I (Day 1–Day 7). Patients who completed the open-label run-in were assessed and randomized (1:1 ratio) under double-blind conditions on Day 0 to receive either AP or placebo tablets for 1 week.
Treatment II (Day 8–Day 14). As part of the crossover design, patients were assessed under double-blind conditions on Day 7 and received for 7 days the test medication in the order of Treatment II, which is the one they did not receive during Treatment I.
Treatment III (Day 15–Day 21). After being assessed on Day 14, patients went back to the same test medication as in Treatment I, beginning on Day 15 for 7 days.
Randomization of the patients through Treatments I–III created the two arms APA and PAP.
Follow-up and termination visit (Day 21–Day 28). Patients withdrew from the study medication/placebo after the assessment on Day 21 and entered a week of follow-up. On Day 28 (±1 day), the termination visit assessment was carried out.
Withdrawal from treatment demonstrated a return to baseline disease state, requiring modification of the ongoing therapies.
Outcomes
Safety endpoint
The primary safety endpoint of the study was to characterize the overall safety and tolerability of AP compared with placebo in patients with MuSK-MG.
Adverse event (AE) records were collected at each visit. Vital signs, complete physical examinations, and electrocardiograms were conducted at screening, at Day 0, and at each visit during the randomization period. Clinical laboratory tests, which included serum chemistry, hematology, and urinalysis, were collected at the same time points.
Efficacy endpoints
The co-primary efficacy endpoints were the differences between AP-treated patients compared with placebo-treated patients in change from baseline in (1) Quantitative Myasthenia Gravis score (QMG) 18 and (2) Myasthenia Gravis–specific Activities of Daily Living Profile (MG-ADL) score. 19
Secondary efficacy endpoints were the differences between AP-treated patients compared with placebo-treated patients in change from baseline in (1) MGC scale score, 20 (2) Carlo Besta Neurological Institute–Myasthenia Gravis (CBNI-MG) scale score, 21 (3) Myasthenia Gravis Quality of Life—15 questions (MG-QoL 15) score, 22 and (4) Fatigue Severity Scale (FSS) score. 23 Baseline was defined as the results of patient evaluations on the last day of open-label run-in, just prior to randomization.
The MG-scale assessment was performed each time starting 45 min after the last dose of medication, following the sequence as listed: MGC, CBNI-MG, QMG, FSS, MG-QoL 15, and MG-ADL.
Randomization and blinding
The randomization list of the study was generated by an independent biostatistician (SPARC Consulting) using the procedure PROC PLAN of the SAS® System version 9.2 software. Randomization was performed by a block randomization method where the two arms of treatments (APA and PAP) were balanced, and the block size for each arm was 10 patients.
The crossover design had each patient receive both active and placebo treatments, falling in one of the two study arms (APA or PAP) according to the randomization. The test medications for the different arms of treatment were dispensed to patients by a trained site pharmacist, according to the randomization schedule, and by the order of randomization number reported on the labeled boxes containing the two treatments (APA and PAP). The study drug and placebo appeared identical. Both the patients and investigator were blinded to treatment assignment.
Statistical methods
Sample size determination
The sample size for this study was based on clinical considerations related to the epidemiology of the disease and not on a formal statistical power calculation.
Since the primary endpoint was safety, we considered that 20 patients could be a reasonable number for such a rare clinical condition and in consideration that the AP safety has been already investigated in patients affected with LEMS.
Safety analysis
Safety analysis was conducted on the safety population (i.e. all randomized patients who receive at least one dose of AP or placebo). The safety analysis is presented using descriptive statistics. Treatment-emergent adverse events (TEAEs) are included in the safety analysis. The incidence of TEAEs is summarized by system organ class, preferred term, relationship to treatment, and severity by treatment group.
Efficacy analysis
Efficacy analysis was conducted on all randomized patients who received at least one dose of AP (AP or placebo) and had at least one post-treatment efficacy assessment.
Differences between AP and placebo in the QMG, MG ADL, MGC, CBNI-MG, MG-QoL 15, and FSS total scores were analyzed using a switchback model for three-period, two-treatment crossover design. The possible carryover effect between AP and placebo was also analyzed by means of classic crossover and one-way analysis of variance (ANOVA) or parallel design analysis models. Least-squares (LS) means and standard errors (SEs) were presented for all efficacy outcome measures in AP and placebo groups.
Concordance between the different evaluation scales was evaluated using Cohen’s kappa coefficient (κ). We first calculated the differences in total scores of each outcome measures to each time point from baseline. Second, we assigned each patient in “improved” or “not improved” categories using different deltas: change of 2, 3, or 4 points from baseline. After that, we calculated the concordance between every combination of these classes and we chose the better result for each outcome measure.
Statistical analyses were performed using the statistical software package SAS v9.4. All significance testing was two-tailed at a 0.05 significance level.
Results
Patients
A total of 20 patients were originally planned to be enrolled for this study between February 2016 and June 2017, but the recruitment was halted after 10 patients because there was a clear beneficial effect on muscle strength and fatigability due to one of the two treatments. All patients included in MuSK-001 trial were enrolled at Istituto Neurologico Carlo Besta. The first informed consent was signed on 19 February 2016 and the last one on 21 November 2016. All patients were in class IIIb or IVb of the MGFA clinical classification representing, respectively, moderate and severe forms of MG predominantly affecting bulbar muscles and, less or equally, limb and axial muscles. Of the 10 patients, 3 were not randomized since they did not reach a stable, therapeutic dose during the run-in period. Following discontinuation, the three patients underwent PE treatment and received immunosuppressants or steroid treatment.
At randomization, four patients were assigned to PAP arm of treatment and three patients to the APA arm (Figure 2). Descriptive statistics about baseline demographic characteristics regarding age of patients, gender, age at onset, and MG duration are reported in Table 1. Overall, there was a female prevalence (85.7% of the entire safety population; 100% of the PAP arm, 66.7% of the APA arm) and a mean age of 43.4 (SD = ±7.0) years, with a mean age at onset of the MusK-MG of 37.0 (SD = ±11.2) years. Five (71.4%) of the seven patients were receiving treatments for MuSK-MG. Steroids were received by three patients, all belonging to the APA group, and one patient in the same group was also receiving mycophenolate mofetil (33,3%). Two patients in the PAP group received azathioprine (25%) and methotrexate (25%), respectively. None of the patients was previously exposed to AP or a closely related drug.
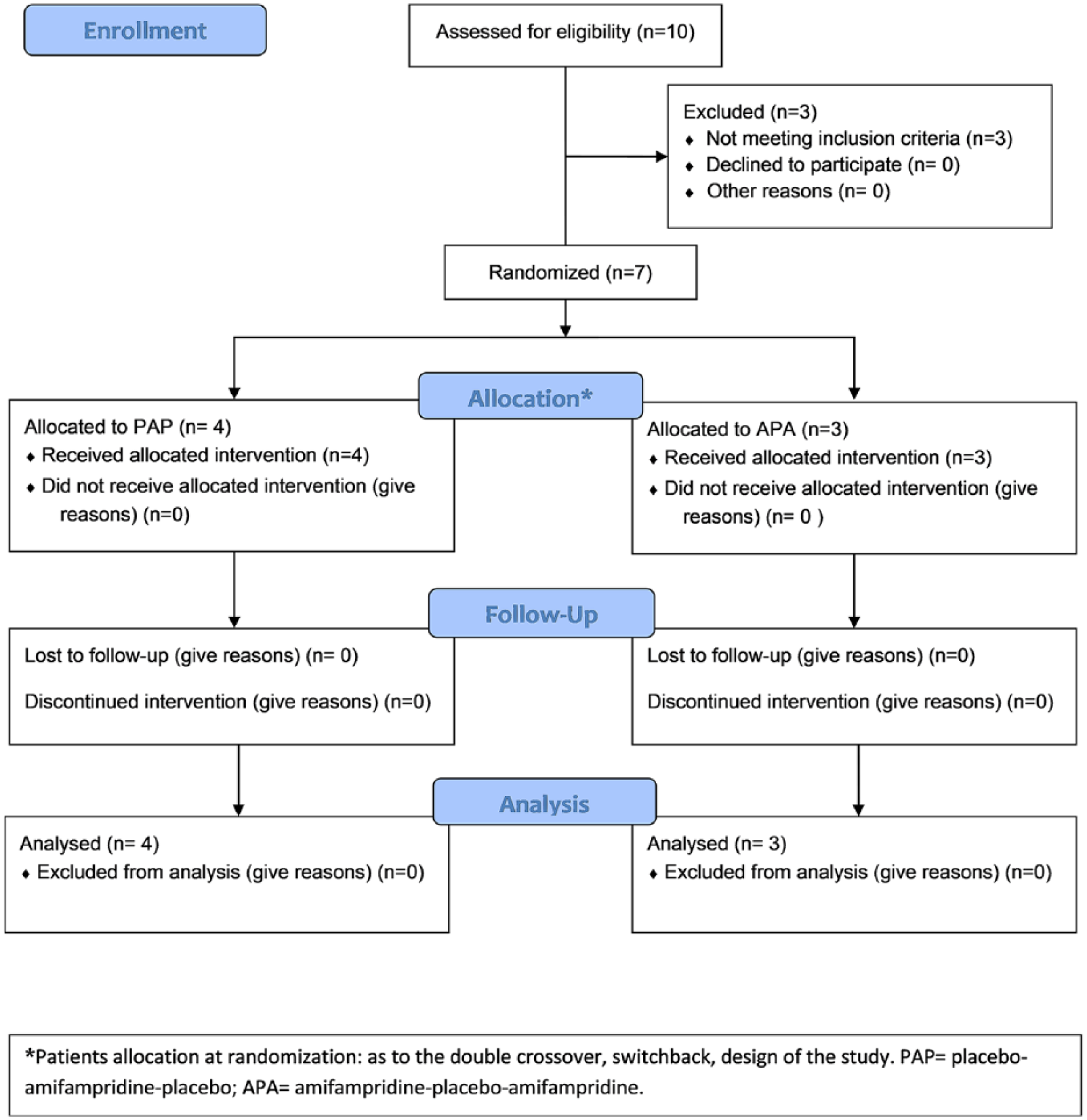
Patients’ disposition diagram.
Baseline demographics of patients recruited to MuSK-001.

PAP: placebo–amifampridine–placebo sequence; APA: amifampridine–placebo–amifampridine sequence; SD: standard deviation.
During the run-in period, the AP effective dose was achieved for all the patients in the range of 30–60 mg daily, with a mean daily dose for AP equal to 47.1 ± 11.1 mg. In particular, one patient (14.3%) maintained the 30-mg starting dose and two patients (28.6%) were assigned to each dosage of 40, 50, and 60 mg.
The overall treatment compliance during the three double-blind periods was 99.6% in the AP group and 94.7% in the placebo group.
Primary safety endpoint
During the run-in period, a total of six patients (60%) reported limbs and/or oral paresthesias, with resolution in a few days (mean = 9.5 days, range = 5–15 days). Two (28.6%) of these six patients experienced intermittent paresthesias after randomization. The reported AEs were considered mild and related to the study treatment. One patient (14.3%) reported chalazion, corneal abrasion, and cystitis after randomization; these AEs were graded as mild and considered unrelated to AP treatment. No serious AEs were reported in the study population, either in the run-in or randomization phase. No patients discontinued treatments because of AEs. No relevant differences between the two groups for hematological tests and blood chemistries, vital signs, and electrocardiogram findings were found over the duration of the study.
Efficacy endpoints
The efficacy endpoints included both patient-reported outcomes (PRO) and physician-reported outcomes (PhyRO). PRO comprised MG-ADL, MG-QoL 15, and FSS, whereas PhyRO consisted of QMG, MGC, and CBNI-MG scales. The primary efficacy measures (MG-ADL and QMG) reflect both patient- and physician-based outcomes. Table 2 presents the change from baseline LS mean scores, as overall mean of all treatment periods post randomization, for AP and placebo; the LS mean difference (95% confidence interval (CI)) between the treatment groups; and the respective p-values. Notably, there was a statistically significant worsening in all the assessments, from baseline, in patients not receiving AP.
Amifampridine versus placebo effect in MuSK-MG patients using different evaluation scales.
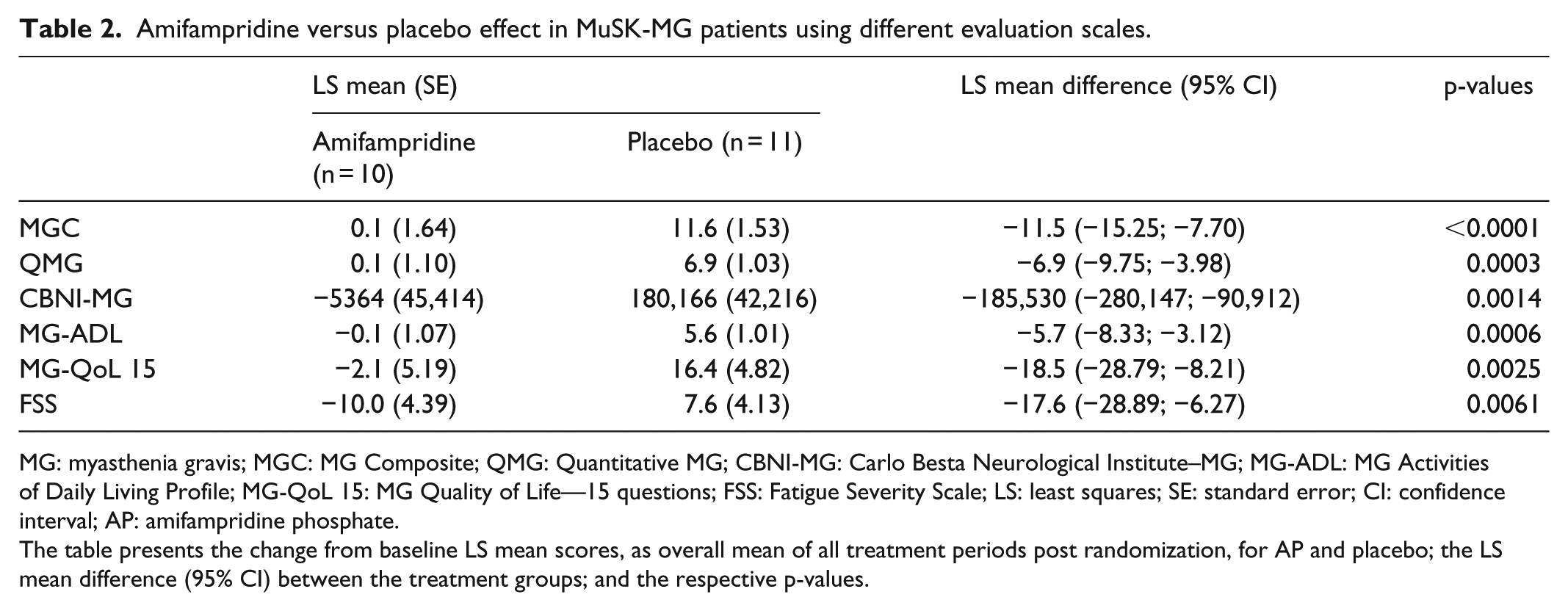
MG: myasthenia gravis; MGC: MG Composite; QMG: Quantitative MG; CBNI-MG: Carlo Besta Neurological Institute–MG; MG-ADL: MG Activities of Daily Living Profile; MG-QoL 15: MG Quality of Life—15 questions; FSS: Fatigue Severity Scale; LS: least squares; SE: standard error; CI: confidence interval; AP: amifampridine phosphate.
The table presents the change from baseline LS mean scores, as overall mean of all treatment periods post randomization, for AP and placebo; the LS mean difference (95% CI) between the treatment groups; and the respective p-values.
Agreement range among the different scales used to evaluate the clinical efficacy of AP was fair to almost perfect, as measured by Cohen’s kappa coefficient. Figure 3 illustrates the concordance κ values between the different PhyRO/PRO evaluations, with MGC at 3 point difference or MG-ADL at 2 point difference as the reference values, respectively (panels A and B). The p-values were all highly significant, suggesting that patients’ perception of muscle strength and fatigability improvement was concordant with that of the evaluating neurologists. In particular, delta of 3 points in QMG had high concordance with delta of 3 points in MGC (κ = 0.81, p < 0.001) and delta of 4 points in QMG had high concordance with delta of 2 points in MG-ADL (κ = 0.81, p < 0.001).
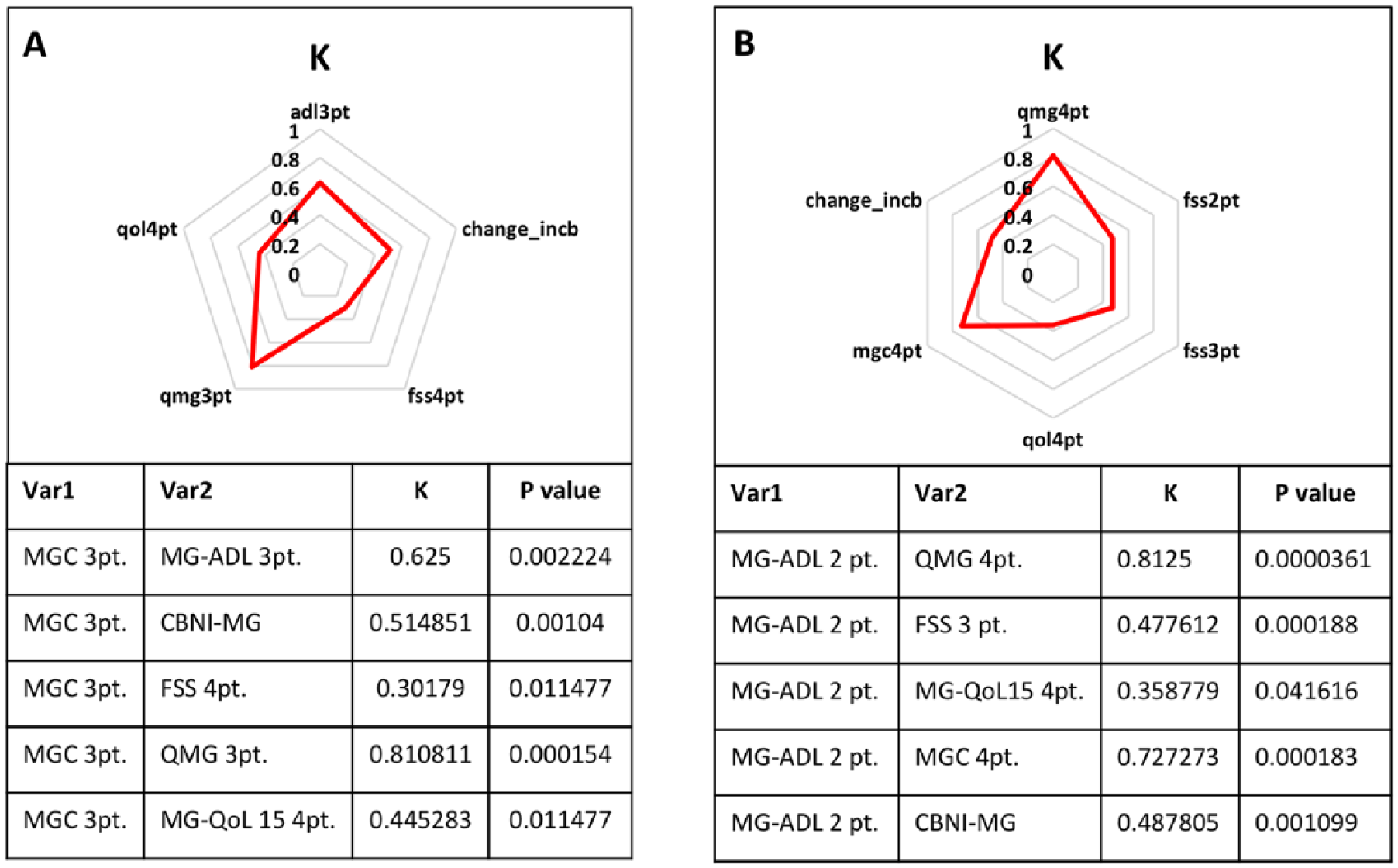
Concordance κ values among the different evaluation scales in AP-treated MuSK-MG patients having MGC (panel A) and MG-ADL (panel B) as a reference evaluation.
Discussion
MG patients with MuSK antibodies represent a subgroup of patients characterized by a severe form of the disease, with a predominance of bulbar symptoms, and resistance to standard treatment. 3 In MuSK-MG, where the balance between MuSK and ACh signaling is disrupted, cholinesterase inhibitors exacerbate the MuSK antibody-induced impairment at the NMJs, leading to neuromuscular hypersensitivity and cholinergic symptoms.4,9 Therefore, a drug which improves neuromuscular transmission in MuSK-MG fulfills a true medical need. In this regard, a strong body of preclinical2–5,9–11,15 and clinical2,6–8,12–14,16–18,24 evidences suggests AP as a good option for the treatment of MuSK-MG patients.
In this study, we demonstrate for the first time the safety and efficacy of AP in MuSK-MG patients in a monocentric, crossover, placebo-controlled, phase IIb trial.
The crossover design was selected because of the following advantages: (1) the decrease in confounding covariates’ influence, since each crossover patient serves as his or her own control; (2) the statistical efficiency of crossover designs, which allows the enrollment of fewer subjects, a relevant need in case of a rare disease such MuSK-MG.25,26
The original target of the study was the recruitment of 20 patients. However, due to the difficulty in recruiting from a single center and an apparent clinical effect, the trial was terminated early, when 10 patients were enrolled. Moreover, the decision to stop study recruitment was taken for ethical reasons because the degree of clinical improvement did not justify the continuation of a pilot study with a placebo arm.
Despite the small number of patients, the trial reported significantly positive results as both the primary and secondary endpoints were met. This was particularly relevant from a clinical standpoint considering that all recruited patients were affected with bulbar symptoms (Table 2). Overall, the side effects were minimal and there were no study withdrawals because of AEs.
An analysis of the concordance between the assessment scales using Cohen’s coefficient indicated that the evaluation of the clinical improvement obtained through the patient point of view—i.e. MG-ADL, MG-QoL 15, and FSS—was well correlated with that of the evaluating physician—i.e. QMG, MGC, and CBNI-MG (Figure 3). Furthermore, the analyses of the MG-ADL, MGC, and QMG scores completely correlated with the trend of assumption of AP versus placebo, at the single patient level, along the different phases of the trial arms (Figure 4), thus strengthening our observations on the efficacy of AP versus placebo. No carry-over effect of the drug was detectable during the different periods of randomization in MuSK-001 trial; indeed, the clinical improvement, or worsening, was rapidly detected by the evaluators and strictly associated with AP administration. Speed of improvement or worsening, occurring within the next day of drug change (AP or placebo), negated an “order effect” as well as a “learning effect,” confirming the fast action of AP as a symptomatic treatment.

Change from the baseline of MG-ADL, MGC, and QMG scores in the single MuSK patients treated with AP.
A clear limitation of the current study is the small sample size. However, the adoption of a switchback, crossover protocol allowed an increase in the number of observations that ensured the statistical power to accomplish a proof-of-concept RCT.
Conclusion
MuSK-001 represents the first double-blind, placebo-controlled, proof-of-concept trial of AP in patients affected by MuSK-MG. This study demonstrated the safety of AP and a statistically significant difference across multiple efficacy assessments, both in the primary and secondary endpoints. Based on the results of this study, a larger multi-center trial is warranted to confirm these results.
Footnotes
Acknowledgements
The authors wish to thank Catalyst Pharmaceuticals for supplying amifampridine phosphate and placebo and for partially funding this no-profit clinical study. They also wish to thank the Italian Association for Myasthenia Gravis (AIM) for supporting patients and promoting clinical research in MG.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical approval
Ethical approval for this study was obtained on 14 July 2015 from the Ethical Committee of the Istituto Neurologico Carlo Besta (approval number: 20/21H).
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Catalyst Pharmaceutical partially funded this no profit clinical study.
Informed consent
Written informed consent was obtained from all subjects before the study.
Trial registration
Italian Agency for Drug (AIFA) Observatory identifier (46/2015).