
Abstract
Pathogenic variants in EEF1A2, a gene encoding a eukaryotic translation elongation factor, have been previously reported in pediatric cases of epileptic encephalopathy and intellectual disability. We report a case of a 17-year-old male with a prior history of epilepsy, autism, intellectual disability, and the abrupt onset of choreo-athetotic movements. The patient was diagnosed with an EEF1A2 variant by whole exome sequencing. His movement disorder responded dramatically to treatment with tetrabenazine. To the best of our knowledge, this is the first report of successful treatment of a hyperkinetic movement disorder in the setting of EEF1A2 mutation. A trial with tetrabenazine should be considered in cases with significant choreoathetosis.
Introduction
De novo variants in EEF1A2 are associated with early infantile epileptic encephalopathy 33 (MIM# 616409) and autosomal dominant intellectual disability 38 (MIM# 616393). A de novo EEF1A2 variant, p.G70S, was initially reported by De Light et al. 1 in a female with epileptic encephalopathy. Subsequently, 16 additional patients have been described in the published literature with 10 different de novo variants. Consistent clinical features include epilepsy/epileptic encephalopathy and global developmental delay/intellectual disability of varying severity (see Table 1). Autism or autistic features, behavioral abnormalities such as stereotypies or self-injurious behaviors, cerebral atrophy, microcephaly, and facial dysmorphism have also been reported in some patients. A recent conference report described abnormal movements in 14 patients with de novo variants in EEF1A2. 9 Patients were noted to have focal or generalized epilepsy, chorea and/or dystonia, and developmental encephalopathy with neuroimaging features of global cortical volume loss and hypomyelination.
Summary of cases.
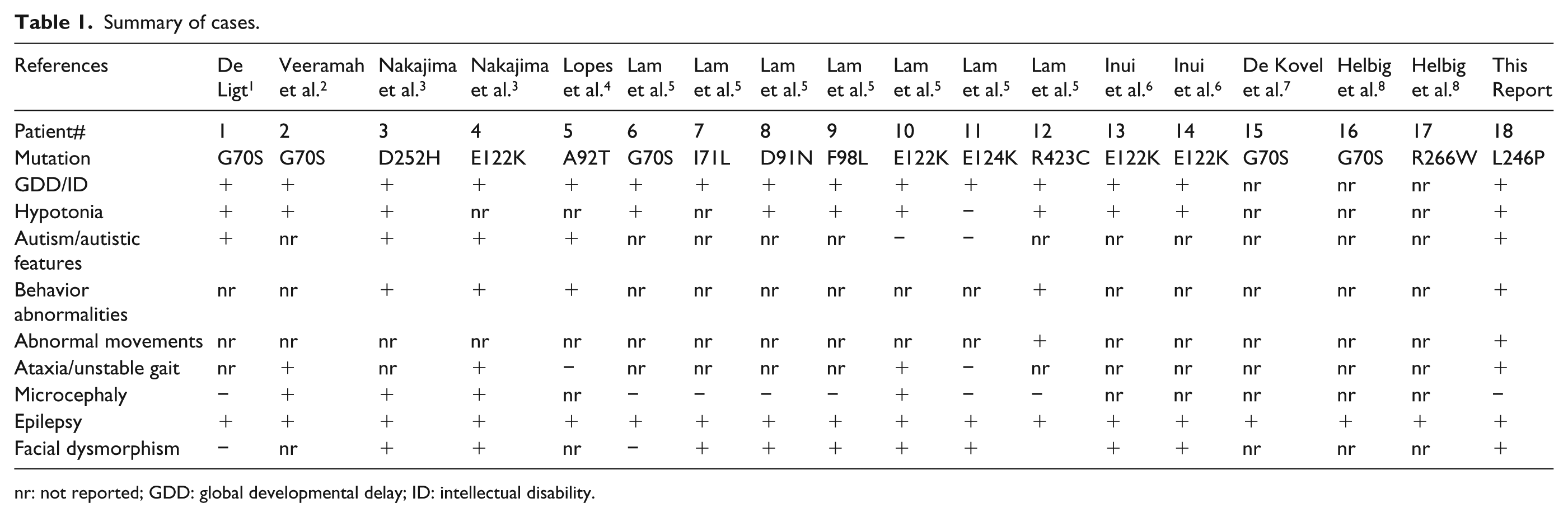
nr: not reported; GDD: global developmental delay; ID: intellectual disability.
Case
The patient is a 17-year-old male born at term following a pregnancy complicated by maternal alcohol and substance abuse. Development was delayed: he walked at age 18 months, requiring braces for support, and his first words were at age 4 years. Medical history includes strabismus status post surgery at age 1 year, hypothyroidism, asthma, gastroesophageal reflux disease, kidney dysfunction, ischemic stroke, and epilepsy well controlled on valproic acid. The patient had an episode of dystonia associated with anti-psychotic treatment (haloperidol, aripiprazole) for bipolar disorder and aggression at age 9. Limited medical record documentation exists regarding the history of ischemic stroke. Magnetic resonance imaging (MRI) of the brain at ages 9 and 12 were normal. Family history is positive for bipolar disorder in his mother. Paternal family history is unknown. The patient has three healthy maternal half-siblings; ancestry is Caucasian.
He first presented to our institution at age 15 years 6 months for management of his seizures. The patient had a wide-based gait, balance impairment, and dysarthria. He demonstrated developmental skills at the 3- to 5-year level. At 2 months after his initial evaluation, he had the abrupt onset of severe choreo-athetotic movements affecting his face, arms, and trunk (see Supplemental Video). These movements dramatically affected his gait and resulted in use of a wheelchair. His speech, behavior, and short-term memory worsened as well. There was no acute preceding event or change of medication (benztropine 1 mg twice daily, valproic acid 500 mg twice daily, dextroamphetamine 5 mg once daily, diphenhydramine 50 mg every 6 h as needed, levothyroxine 31.25 mcg once daily, fluoxetine 20 mg once daily). Shortly thereafter, the patient was admitted twice to a psychiatric hospital because of anger issues and aggression, but did not require change of medication. He had another seizure after his 16th birthday and a third brain MRI was normal. Over the next several months, his condition worsened due to the involuntary movements and he was no longer able to feed or bathe himself. Gastrostomy (G) tube feeding was required for caloric support. His hyperkinetic movements failed to respond to benztropine 1 mg BID and valproic acid 500 mg BID.
Evaluation in a Pediatric Movement Disorders clinic at age 16 years and 4 months showed continuous choreo-athetotic movements of his face, trunk, and extremities. A trial with tetrabenazine (TBZ) 12.5 mg BID was initiated, which markedly improved his involuntary movements within 2 weeks. At his most recent follow-up visit (age 17 years), he was able to walk, feed himself, and use the bathroom independently. He continued to have antecollis and difficulties with gait, balance, and dysarthria, but was close to his baseline presentation.
Additional neurodiagnostic testing included a normal (fourth) brain MRI, single-nucleotide polymorphism (SNP) chromosomal array, Fragile X DNA testing, and mitochondrial DNA sequencing and deletion/duplication testing. A fasting lumbar puncture revealed a slightly low cerebrospinal fluid (CSF) glucose of 48 mg/dL (normal range 50–75); however, plasma-to-CSF glucose ratio (0.56) was not suggestive of GLUT1 deficiency. CSF lactate, pyridoxal phosphate, neurotransmitter metabolites, tetrahydrobiopterin, neopterin, 5-methyltetrahydrofolate, and succinyladenosine were all normal. CSF amino acids did not identify any abnormalities consistent with a metabolic disorder. Plasma amino acids showed elevated alanine and glycine, but were normal on repeat testing. Lactate, pyruvate, and urine organic acids were normal. In light of extensive negative work-up, whole exome sequencing was performed using the methods previously described. 10
Results identified a heterozygous missense variant, c.737T>C (p.L246P) in the EEF1A2 gene (NM_001958.3). Targeted testing of the patient’s maternal grandmother was negative; biological parental samples were not available to confirm a de novo occurrence in the patient.
Discussion
The defined EEF1A2 abnormalities in this patient have not been previously reported in affected individuals, and it is absent from 138,632 exome/genome sequences in the Genome Aggregation Database (http://gnomad.broadinstitute.org/). The p.L246P variant is a semi-conservative amino acid substitution that occurs at a position that is evolutionarily conserved across species, and in silico analysis predicts that this alteration is likely damaging to protein structure/function. 11 It is located in domain II of the protein, which is involved in the binding of aminoacyl-tRNA. Pathogenic variants have been documented at nearby residues (e.g. p.D252H), 3 supporting the functional importance of this region. We interpret this variant to be causative of the patient’s neurodevelopmental impairments, epilepsy, facial dysmorphism, and movement disorder.
The EEF1A2 gene encodes the eukaryotic translation elongation factor-1, alpha-2, which is critical for the GTP-dependent binding of aminoacyl-tRNAs to the ribosome during protein synthesis. 12 The genes which encode the two eEF1A isoforms are expressed ubiquitously during early development, but EEF1A1 and EEF1A2 are expressed in mutually exclusive somatic patterns around the time of weaning in juvenile mice. Complete homozygous loss of EEF1A2 expression in mice gives rise to the “wasted” phenotype, in which the affected mice exhibit motor neuron disease with concomitant skeletal muscle atrophy leading to morbidity requiring euthanasia within 1 month of life. 13
A recent article showed that the human pathogenic p.G70S eEF1A2 variant, introduced into mice via CRISPR mutagenesis, mimicked the wasted phenotype with the addition of a fatal audiogenic seizure phenotype. 14 Furthermore, a mouse model which carried two copies of p.G70S exhibited an accelerated wasting phenotype requiring euthanasia at 18 days old. These data suggest that the p.G70S variant might confer a pathologic gain of function in addition to its loss of protein translation function. Nevertheless, it is recognized that the p.G70S mutation is in a physically and functionally remote location on the eEF1A2 protein relative to the p.L246P mutation identified in our patient. It remains unclear whether the neuropathologic phenotypes are a general consequence of human EEF1A2 loss-of-function variants or correlate to mutations in distinct regions of the eEF1A2 protein.
Conclusion
Movement abnormalities associated with variants in EEF1A2 are variable, including chorea, dystonia, unsteady gait, and/or ataxia. Our patient had an early history of ill-defined involuntary movements. Transient dystonic movements were reported with use of dopamine antagonists (age 9), and 6 years later he developed severe choreo-athetotic movements. The latter movements, we believe, are attributed to his underlying genetic mutation, although it is recognized that fluoxetine is associated with hyperkinesia in about 2% of children and adolescents and the patient has preceding history of prenatal drug exposure and reportedly a prior ischemic stroke. As described, his hyperkinetic movements dramatically responded to TBZ. TBZ depletes dopamine by inhibiting the vesicular monoamine transport of this neurotransmitter. TBZ is approved by the US Food and Drug Administration for treatment of chorea associated with Huntington disease; however, it has been shown to be effective for a large variety of hyperkinetic disorders. 15 Practitioners must also consider the potential adverse central nervous system effects of TBZ that might be particularly concerning in patients with epileptic encephalopathy, specifically fatigue, sedation, depression, and falling. Based on this report, a trial with TBZ should be considered in those individuals with significant choreoathetosis and an EEF1A2 mutation.
Footnotes
Declaration of conflicting interests
J.S.C. is a consultant for Invitae. H.S.S. is a consultant for TEVA pharmaceuticals. A.F. has received personal compensation in the form of consultant fees from Aevi Genomic Medicine, Vertex Pharmaceuticals, Stealth Biotherapeutics, BluebirdBio, and Calico Labs for services unrelated to the reported topic. The other authors declare that they have no competing interests.
Ethical approval
Our institution does not require ethical approval for reporting individual cases or case series.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Informed consent
Informed consent was obtained from the subject’s legal guardian to publish this case report and related video.
Supplemental material
Supplemental material is available for this article online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.