
Abstract
Atherosclerosis is a chronic inflammatory disease affecting the vascular system, characterised by the accumulation of modified lipoproteins, immune cell aggregation and the development of fibrous tissue within blood vessel walls. As atherosclerosis impacts blood vessels, its adverse effects may manifest across various tissues and organs. In this review, we examine the association of atherosclerosis with Alzheimer’s disease, stroke, pancreatic and thyroid dysfunction, kidney stones and chronic kidney diseases. In several cases, the reciprocal causative effect of these diseases on the progression of atherosclerosis is also discussed. Particular attention is given to common risk factors, biomarkers and identified molecular mechanisms linking the pathophysiology of atherosclerosis to the dysfunction of multiple tissues and organs. Understanding the role of atherosclerosis and its associated microenvironmental conditions in the pathology of multi-organ disorders may unveil novel therapeutic avenues for the prevention and treatment of cardiovascular and associated diseases.
Keywords
Introduction
Atherosclerosis is a chronic inflammatory disease and the primary driver of cardiovascular diseases (CVD), ultimately leading to myocardial infarction and stroke, and significantly contributing to mortality in Western nations. 1 The pathogenesis of atherosclerosis is conventionally divided into distinct ‘stages’ (e.g. initiation, progression and advanced stages with complications). Hypertension, hyperglycaemia, dyslipidaemia, diabetes mellitus, physical inactivity, obesity, poor nutrition, stress, environmental factors and genetic predisposition are recognised as major risk factors associated with atherosclerosis. 2 Moreover, atherosclerosis often coexists with a related condition known as arteriosclerosis. While atherosclerosis refers to the deposition of fibro-fatty lesions in arterial walls, arteriosclerosis is characterised by stiffening of the arterial media due to connective tissue degeneration, particularly of its main component, elastin. Unlike atherosclerosis, arteriosclerosis is considered a consequence of systolic hypertension resulting from aortic stiffening and other adverse haemodynamic effects. 3
The initiation of atherosclerosis is characterised by the excessive accumulation of immune-reactive, multiple modified forms of low-density lipoprotein (LDL) particles (modified by glycation, oxidation, desialylation, dicarbonylation and other processes). These modifications induce inflammation, promote foam cell formation and stimulate both humoral and adaptive immunity.4 –6 Thus, inflammation, lipid accumulation and oxidative damage are key processes involved in the initiation of atherosclerosis. As the disease progresses, the formation of a necrotic plaque core and the onset of mineralisation occurs. While extensive calcification can stabilise plaques, spotty calcification is known to reduce plaque stability, increasing the risk of rupture.7,8 In the advanced stage, the growing atherosclerotic plaque disrupts normal blood flow and limits oxygen supply to tissues and organs (ischaemia). 9 Advanced plaques are also prone to rupture, triggering acute thrombosis in blood vessels and causing myocardial infarction or stroke. 10
As the vascular system is integral to all tissues and organs, atherosclerosis impacts the functionality of the entire body, extending beyond the typical anatomical sites (e.g. thoracic aorta, aortic valve or coronary artery) where atherosclerotic plaques are usually studied. The association of atherosclerosis with pathologies in numerous tissues and organs is well-documented and has been discussed extensively in recent reviews: heart,11,12 liver,13 –15 immune system,16 –19 bone marrow,20,21 lymph nodes and lymphoid organs,22 –24 adipose tissue,25,26 vascular smooth muscle cells,27 –29 skin,30 –32 gastrointestinal tract33,34 and pancreas (in the context of diabetes).35,36 Therefore, these topics will be excluded from this article. In this review, we focus on the association of atherosclerotic cardiovascular disease (ASCVD) with selected major organ systems and specific diseases: the brain (ischaemic stroke (IS) and Alzheimer’s disease (AD)), pancreas (beyond diabetes), kidneys (cholesterol crystal embolism (CCE), chronic kidney disease (CKD) and kidney stone (KS) disease) and thyroid dysfunction (hypo- and hyper-thyroidism). Our goal is to provide an updated conceptualisation of how atherosclerosis is implicated in and associated with these multi-organ pathologies, expanding our understanding of the underlying molecular mechanisms and offering new insights for the development of therapeutic opportunities.
Association between ASCVD and IS
IS, a cerebrovascular accident, is the second leading cause of morbidity and mortality worldwide, with over 13.7 million cases identified annually. 37 Atherosclerosis plays a crucial role in the pathogenesis of IS, characterised by vascular dysfunction and either significant stenosis or occlusion of a major brain artery (carotid or vertebral arteries) or a branch of a cortical artery (anterior, middle or posterior cerebral arteries), resulting in hypoxia-induced neuronal injury. 38 Other aetiological subtypes of IS include cardioembolism, small vessel occlusion, stroke of other aetiology (e.g. dissections or vasculitis) and stroke of undetermined aetiology. 39
Atherosclerosis and stroke share several risk factors, such as hypertension, type 2 diabetes (T2D) and metabolic syndrome, further highlighting the close association between these pathologies (Figure 1). 40 For instance, a recent study involving 23,973 Chinese participants with no history of CVD demonstrated that systolic blood pressure (BP) had a stronger association with IS than plaque burden. While plaque burden was not linked to probable cardioembolic stroke, it showed a stronger association with probable large artery stroke compared to lacunar stroke. 41

Potential pathophysiological mechanisms connecting atherosclerosis and stroke thought the common risk factors (diabetes, metabolic syndrome and hypertension). Oxidative stress and chronic low-grade inflammation are the main processes leading to endothelial dysfunction and arterial stiffness, and, eventually, resulted in atherosclerosis. Subsequently, atherosclerosis progression may lead to thrombosis, formation of unstable plaques and stenosis of blood vessels, which greatly increase the risk of stroke and other cardiovascular events. Post-stroke dementia is a common condition of stroke survivors, seen at a high rate shortly after stroke.
Moreover, many markers associated with atherosclerosis – such as biochemical parameters, inflammatory molecules, genetic factors and RNAs (micro-, circular-, long non-coding- and transfer RNA-derived small RNAs) – serve as prognostic indicators of mortality in stroke patients. 42 For example, C-reactive protein (CRP), expressed in vascular smooth muscle cells and implicated in plaque destabilisation, has been recognised as an independent predictor of mortality in stroke patients, as well as for the risk of recurrent stroke. 43 Additionally, the recently identified Embryonic Lethal Abnormal Vision Drosophila-like protein 1 (ELAVL1) is associated with the development of CVD and cerebral ischaemia/reperfusion injury. 44 Serum ELAVL1 levels are higher in IS patients compared to carotid atherosclerosis patients and correlate positively with total cholesterol (TC), LDL-C, CRP and pro-inflammatory cytokines (tumour necrosis factor-α (TNFα) and interleukin-6 (IL-6)), while negatively correlating with high-density lipoprotein-cholesterol (HDL-C). These findings suggest that serum ELAVL1 could serve as a biomarker for diagnosing IS. 45
Migraine, a complex neurological disorder characterised by episodic headaches, is a well-established risk factor for myocardial infarction, total haemorrhagic and IS.46 –48 However, migraine is not associated with increased atherosclerosis in large vessels in acute ischaemic stroke (AIS) patients. On the contrary, studies suggest a lower prevalence of atherosclerotic changes in stroke patients with migraine. This indicates that the increased risk of IS in migraine patients may be based on pathological mechanisms other than atherosclerosis, warranting further investigation. 49
Modern imaging techniques, such as computed tomography (CT), are widely used to diagnose both atherosclerosis and stroke in clinical settings by evaluating plaque properties and calcification characteristics.50,51 For example, the high prevalence of napkin-ring sign plaques observed in cervicocerebral CT angiography has been proposed as a significant risk factor for AIS. This biomarker may be particularly valuable for routine and emergency screening of asymptomatic atherosclerotic patients to identify their risk of AIS and apply timely anti-atherosclerotic therapies for prevention. 52
Vascular calcification, characterised by calcium–phosphate complex deposition in vessels, affects plaque stability (intimal calcification) and causes arterial stiffening (medial calcification). Among others, carotid artery calcification, intracranial artery calcification and coronary artery calcification are the most studied quantitative parameters for predicting the risk of atherosclerosis and stroke.53,54 For instance, in AIS patients undergoing intravenous recombinant tissue plasminogen activator (thrombolysis) treatment, the total carotid siphon calcification score (8-point scale) was associated with mortality within the first 3 months. 55 Additionally, calcification volumes in major vessels (e.g. intracranial internal carotid artery, cervical carotid artery and aortic arch) were higher in AIS patients aged < 65 years with large artery atherosclerosis compared to other stroke aetiologies. 56 The specific “spotty” calcification pattern (small, scattered calcium deposits) has been linked to high-risk lesions. 57 Notably, recent research indicates that the prevalence of spotty calcification at the carotid bifurcation and siphon is associated with an increased risk of non-lacunar IS compared to control patients with subclinical atherosclerosis. 58 Interestingly, a study of 207 AIS patients revealed that high serum aldosterone levels independently predicted advanced intracranial arterial calcification and atherosclerosis. 59 These findings suggest that aldosterone, a key mineralocorticoid steroid hormone involved in atherosclerotic lesion progression, 60 may serve as a biomarker for atherosclerosis, arterial calcification and stroke risk. 59
Stroke survivors often experience post-stroke cognitive impairment (PSCI), ranging from mild cognitive decline to severe post-stroke dementia. 61 Among AIS patients, both low and high systolic BP were associated with an increased risk of PSCI at 3 months. Additionally, large artery atherosclerosis and total anterior circulation infarct were linked to higher PSCI risk. These findings suggest that maintaining optimal BP levels may help reduce PSCI occurrence. 62 In asymptomatic patients with significant carotid stenosis, the instability of atherosclerotic plaques at the carotid bifurcation was associated with subclinical microemboli and white matter hyperintensities, correlating with vascular cognitive decline. 63 Furthermore, a study involving 128 stroke patients found that progression and multiple calcifications of carotid artery plaques were associated with accelerated PSCI, whereas the absence or single calcifications were not. 64
Atherosclerosis significantly increases the risk of IS through mechanisms involving plaque formation, rupture and embolism. The presence of napkin-ring sign plaques, a ‘spotty’ pattern of plaque calcification, and elevated biomarkers such as aldosterone, CRP and ELAVL1 protein strongly correlate with stroke risk. These findings emphasise the need for early identification and management of atherosclerosis, especially in individuals at high risk for stroke. Stroke biomarkers and plaque characteristics could serve as key indicators for better prevention and treatment strategies.
Association between ASCVD and AD
AD is a progressive neurodegenerative condition characterised by the deposition of amyloid β (Aβ) and hyperphosphorylated tau protein aggregates in the brain, leading to significant cognitive decline. 65 Both AD and atherosclerosis have been identified as chronic low-grade inflammatory diseases that share common risk factors, such as ageing, hyperlipidaemia, vascular dysfunction and hypertension. These shared characteristics suggest a bidirectional relationship wherein each condition may promote the other’s progression (Figure 2). 66

Interplay between cardiovascular risk factors and increased risk of Alzheimer’s disease. Hypertension can increase β-amyloid (Aβ) deposition and aggravate Aβ-induced cerebrovascular dysfunction. Also, hypertension could impair Aβ vascular clearance and increase its cleavage from the amyloid precursor protein (APP). Finally, hypertension can cause white matter injury, microbleeds, microinfarcts and facilitate development of cerebral atherosclerosis, which manifests by ischaemic stroke, chronic hemispheric hypoperfusion or cerebral hypoxia, thus triggering AD development. Diabetes and metabolic syndrome associated with AD through several underlying mechanisms, such as hyperglycaemia-induced toxicity, advanced glycation end product-induced adverse effects, insulin resistance, insulin receptor impairment, inflammation and cerebrovascular damage. Also, dyslipidaemia increased Aβ peptide deposition, promotes Tau hyperphosphorylation, compromised the integrity of the blood–brain barrier and promote neuroinflammation compatible with AD. The role of ASCVD in AD progression is depicted in solid lines; the role of AD in the progression of ASCVD is depicted in dashed lines.
The role of AD in atherosclerosis progression
The amyloid precursor protein (APP) and Aβ are produced in various tissues and organs outside the central nervous system, including the heart, endothelium, intestine, skin, muscle and adipose tissue.67,68 Despite APP’s documented expression in endothelial cells and cardiomyocytes, its physiological and pathological roles in the cardiovascular system remain poorly understood. 69 Cohort-based analyses have demonstrated associations between elevated amyloid-beta 1-40 (Aβ40) levels and increased arterial stiffness, subclinical atherosclerosis and coronary heart disease (CHD), suggesting that Aβ may contribute to pathologies beyond AD. 70 Furthermore, APP overexpression has been shown to exacerbate endothelial dysfunction and atherosclerosis in ApoE−/− mice, while APP deletion reduced atherogenesis in the same model.71,72
Aβ-mediated oxidative damage, lipid oxidation and modification have been proposed as key mechanisms in the initiation of atherosclerosis. 73 For instance, the increased expression of chitinase-3-like protein 1 (Chi3l1) has been observed in the aortas of atherosclerotic patients 74 and in the brains of human APP-expressing mice. 75 High serum Chi3l1 levels have been linked to endothelial dysfunction and thromboembolic stroke.76,77 Knockdown of Chi3l1 in ApoE−/− mice suppressed the progression of atherosclerotic plaques. 74 It has also been shown that APP reduces the expression of miR-342-3p in arterial endothelium, thereby increasing Chi3l1 levels and promoting atherosclerosis. 78 These findings suggest that Chi3l1 is a critical pro-atherogenic factor, while miR-342-3p functions as an anti-atherogenic miRNA regulated by APP. This pathway holds potential as a novel diagnostic and therapeutic target for atherosclerosis.
Another mechanism implicates APP in platelet adhesion and thrombus formation. Platelets from APP knockout mice fail to adhere to immobilised Aβ peptides Aβ1–40, Aβ1–42 and Aβ25–35. In contrast, blood from wild-type mice enhances adhesion to Aβ peptides co-coated with collagen. Aβ also promotes platelet aggregation and degranulation, further contributing to plaque progression and vascular complications.79 –81
The role of atherosclerosis in AD progression
Atherosclerosis has also been identified as a predictive factor for accelerated cognitive decline in AD patients. Clinical and neuropsychological evaluations have revealed that increased carotid intimal medial thickness (IMT) correlates with faster deterioration in memory, executive function and semantic fluency in AD patients. 82 Hypertension serves as a convergence point linking atherosclerosis and AD, acting as a well-recognised risk factor for both conditions.83,84
Experimental models have further elucidated this connection. In TgSwDI mice (elevated Aβ levels only in the brain) and Tg2576 mice (elevated Aβ levels in both blood and brain), acute angiotensin II (ANGII) treatment aggravated cerebrovascular dysfunction exclusively in TgSwDI mice. ANGII was shown to enhance β-secretase activity, increasing the production of toxic Aβ1–42 while reducing Aβ1–40 levels. This pathway highlights the role of hypertension in APP processing and amyloidogenesis, thereby linking vascular dysfunction and AD progression. 85 Clinical studies have supported these findings, showing that hypertension promotes intracranial atherosclerosis, leading to cerebral hypoperfusion, arterial wall stiffening and the progression of AD pathology. 86
High-fat diet-fed Tg mice have demonstrated additional contributions of atherosclerosis to AD pathology, including hypercoagulation, thrombocytosis, chronic platelet activation and memory impairment. Procoagulant platelets facilitate the conversion of soluble Aβ40 into fibrillar aggregates, which obstruct cerebral blood vessels. These changes are accompanied by increased cerebral vascular permeability, heightened neuroinflammation, neuron loss and tau hyperphosphorylation. 87
The CCAAT/enhancer-binding protein beta (C/EBPβ)/asparagine endopeptidase (AEP) pathway has been identified as another shared mechanism between atherosclerosis and AD.88,89 C/EBPβ is a transcription factor regulating inflammation and oxidative stress.90,91 while AEP cleaves APP and tau, accelerating Aβ production and tau aggregation in AD and promoting foam cell formation and vascular remodelling in atherosclerosis.92,93 Knockout studies in Tg and ApoE−/− mice have shown that deletion of C/EBPβ or AEP reduces foam cell formation, arterial macrophage accumulation, cerebral blood flow impairment and cognitive deficits, thereby ameliorating both atherosclerosis and AD pathologies.94 –98
Recent research on Tg and ApoE−/− mice has further explained the C/EBPβ/AEP-mediated association between AS and AD. Therefore, macrophage-specific deletion of C/EBPβ or AEP decreased cholesterol load and reduced foam cell formation and lesions area in aorta of HFD-fed ApoE−/− mice. Knock out of C/EBPβ or AEP from HFD-fed Tg/ApoE−/− mice reduced the lesion area and arterial macrophage accumulation, increased cerebral blood flow and blood vessel length and restored cognitive deficits, thus ameliorating AD pathologies. Knock out of ApoE from hippocampus of Tg mice increased serum LDL and lesion areas in the aorta, reduced cerebral blood flow and vessel length and increased cognitive deficits, thus aggravating AD pathologies. These results showed that C/EBPβ/AEP signalling connected AS to AD through ApoE-mediated cerebral vasculature dysfunctions, where AS-induced brain hypoperfusion contributed to the AD progression. 99
The crucial role of ApoE in both AS and AD progression was shown also on humans, where neuropsychological evaluation of AD patients demonstrated that ApoE ε4 allele was associated with more severe forms of atherosclerosis and higher rate of cognitive decline in AD. 100 AD patients carrying the ApoE ε4 allele and affected by non-valvular atrial fibrillation exhibit the highest IMT, vascular damage and cognitive deficits. 101
These findings suggest that atherosclerosis actively contributes to AD pathology through vascular dysfunction, cerebral hypoperfusion, blood coagulation and platelet-mediated Aβ aggregation, ultimately resulting in cognitive decline. Early detection and timely management of vascular risk factors could prevent or delay the progression of AD, highlighting the importance of integrated therapeutic approaches.
Atherosclerosis and AD are closely interconnected, with vascular dysfunction and inflammation playing central roles in both conditions. The C/EBPβ/AEP signalling pathway and the ε4 allele of ApoE contribute to the progression of both atherosclerosis and AD, with atherosclerosis worsening cognitive decline in AD patients. These findings suggest that targeting atherosclerotic mechanisms may offer therapeutic benefits for AD patients. A dual approach that addresses both cardiovascular and neurodegenerative factors could help slow the progression of both diseases.
Pancreas and ASCVD
The contribution of pancreatic dysfunction and associated metabolic deficiencies, such as insulin resistance and obesity, to the progression of atherosclerosis has been extensively investigated (Figure 3).102 –105 This section explores the association between pancreatic pathologies – particularly pancreatic fat deposition – and atherosclerosis, alongside co-occurring conditions such as T2D and non-alcoholic fatty liver disease (NAFLD).
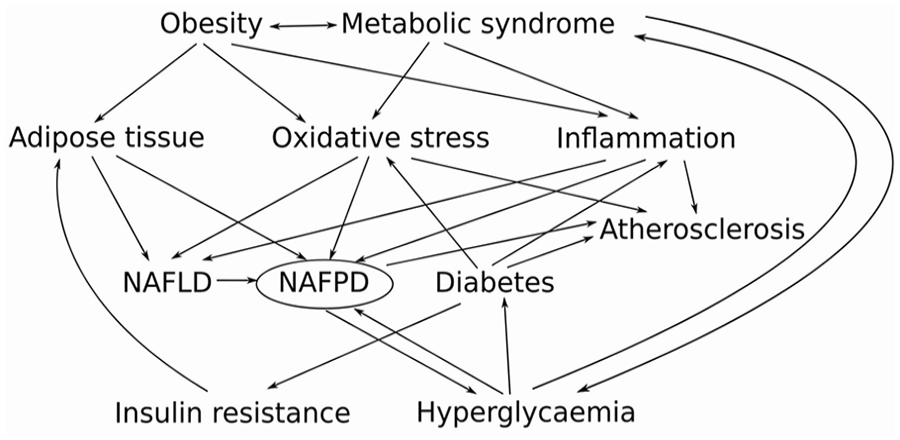
The complex relationship between diabetes, metabolic syndrome, non-alcoholic fatty liver disease (NAFLD), non-alcoholic fatty pancreatic disease (NAFPD), and atherosclerosis. In the conditions of obesity and diabetes, the increased secretion of free fatty acids (FFA) from adipose tissue to the circulation lead to increased delivery of FFA to the pancreas and the liver. In the liver, the surplus of FFA facilitated the development of NAFLD and increased the production of very-low-density lipoproteins, which promoted the development of NAFPD. Also, metabolic syndrome, obesity and diabetes are associated with systemic low-grade inflammation and increased oxidative stress, which, altogether with β-cell lipotoxicity caused by a fat accumulation in the pancreas, leading to the vicious cycle that further aggravates metabolic diseases and NAFPD. Several shared risk factors connected discussed disease with higher risk for the development of atherosclerosis and other cardiovascular diseases.
Fat accumulation in the pancreas has long been recognised and is frequently associated with obesity and NAFLD.106,107 Given the high prevalence of fatty pancreas in the general population, it is now regarded as a common disorder. In one study, 25.9% of subjects undergoing endoscopic ultrasonography were diagnosed with fatty pancreas, which was associated with elevated levels of hyperlipidaemia, aortic IMT, fatty liver frequency, ischaemic heart disease and uric acid levels. 108 Non-alcoholic fatty pancreas disease (NAFPD) has been linked to increased body mass index (BMI), β-cell dysfunction, pancreatic inflammation and fibrosis, all of which can progress to diabetes mellitus, metabolic syndrome or pancreatic cancer.109,110 In patients with NAFLD, high-resolution ultrasonography revealed that 97% exhibited fat deposition in the pancreas. Furthermore, fatty pancreas presence was correlated with increased carotid IMT and carotid-femoral pulse wave velocity, with the grade of fatty pancreas showing a positive correlation specifically with carotid-femoral pulse wave velocity. 111 Another study demonstrated that individuals with NAFPD exhibited higher levels of epicardial adipose tissue and aortic IMT compared to healthy controls. Additionally, NAFPD, age and BMI were independently associated with increased aortic IMT. 112 A large-scale systemic evaluation further found carotid calcification to be more frequent in NAFPD patients, suggesting a potential link between NAFPD and systemic calcified atherosclerosis. 113
Similarly, studies on T2D patients using CT have demonstrated that pancreatic fat deposition correlates with age, visceral fat area and vascular stiffness. Interestingly, the relationship between pancreatic fat and the prevalence of carotid artery plaque or vascular stiffness was only significant in non-obese patients, while in obese patients, no such association was observed. 114 Another study on T2D patients reported a positive correlation between high pancreatic fat levels and carotid plaque formation, regardless of obesity status. 115 Additionally, T2D patients were found to have more extensive splenic artery calcifications and a less-dense pancreatic arterial tree compared to non-diabetic controls. 116
Atherosclerosis contributes to pancreas dysfunction by reducing blood flow, leading to islet hypoxia and β-cell dysfunction, which can result in NAFPD and potentially NAFLD and diabetes. The vascular impact of atherosclerosis on the pancreas further complicates the management of metabolic disorders. These findings suggest that improving vascular health in patients with metabolic diseases, such as diabetes, may help prevent further pancreatic dysfunction. Monitoring blood flow and vascular health could be crucial for early intervention in patients at risk for NAFPD.
Chronic kidney disease
CKD is characterised by the progressive decline of renal function, indicated by a reduction in the glomerular filtration rate over at least 3 months, accompanied by manifestations such as albuminuria and abnormal kidney morphology. CKD is recognised as a prevalent systemic condition, affecting approximately 10% of the global population. 117 The disease is stratified into five stages, with stage 3b marking the irreversible point in disease progression. Patients in stage 5 develop end-stage renal disease (ESRD), characterised by severely diminished kidney function, often necessitating dialysis or kidney transplantation.118,119 Diabetes and hypertension are the primary causes of CKD,120,121 while CCE is frequently overlooked as a cause of CKD in patients with advanced atherosclerosis. 122
Importantly, CKD patients have an elevated risk of developing CVD, making cardiovascular complications, rather than kidney failure itself, the leading cause of mortality in CKD – primarily due to stroke or myocardial infarction.123,124 In addition to traditional risk factors shared by CKD and CVD (such as diabetes mellitus, dyslipidaemia, hypertension, hyperuricaemia, obesity, advanced age, family history, tobacco use and male gender), non-traditional risk factors, including disruptions in calcium-phosphate metabolism, mineral and bone disorders, malnutrition, oxidative stress and uraemic toxins, play a crucial role in CKD-related atherosclerosis development.125 –127 The roles of a disintegrin and metalloproteases (ADAM) 10 and 17 in both CKD and atherosclerosis have been excellently reviewed elsewhere, 128 therefore, will be omitted here.
The role of atherosclerosis in CKD
The rupture of atherosclerotic plaques exposes their core – comprising cholesterol, fatty substances and cellular debris – to the circulation. In the bloodstream, cholesterol crystals (CCs) can become lodged in arterioles, leading to CCE, which narrows or obliterates the arteriole lumen, causing ischaemia and infarction in various tissues and organs, including the brain, muscles, bones, kidneys, nerves, skin, eyes, gastrointestinal tract, visceral organs and extremities. Due to the kidneys’ anatomical proximity to the abdominal aorta and the extent of renal blood flow, cholesterol atheroembolism frequently targets the kidneys, potentially resulting in acute kidney failure.129,130 CCE is now a recognised cause of renal failure in older adults with atherosclerosis. Risk factors for CCE are similar to those for atherosclerosis, including hypercholesterolaemia, hypertension, peripheral vascular disease, diabetes, abdominal aortic aneurysm, ischaemic cardiovascular disease, cerebrovascular disease, hypercoagulability, smoking, male gender, Caucasian ethnicity and other factors. 131
The precise mechanisms underlying CCE remain incompletely understood. CCE is predominantly iatrogenic, often linked to invasive procedures involving the aorta or major arteries, such as coronary angioplasty, angiography or left heart catheterisation.132,133 However, local tissue necrosis, inflammatory responses, activation of the renin–angiotensin–aldosterone system and complement activation triggered by CC are considered critical to CCE development.134 –136
CCE is challenging to diagnose and often heralds severe and unstable atherosclerotic disease, frequently associated with acute cardiovascular events and poor outcomes.
The role of CKD in atherosclerosis development
Comparisons of atherosclerotic parameters between ESRD patients and healthy controls have revealed greater levels of inflammation, vascular dysfunction, malnutrition and calcification in ESRD patients undergoing haemodialysis.137 –139 Elevated levels of von Willebrand factor, a marker of endothelial impairment, have also been observed in CKD and ESRD patients. 140 These findings suggest that CKD-related pathological changes exacerbate vascular conditions, thereby accelerating atherosclerosis development.
The impaired efficacy of glomerular filtration in CKD leads to metabolite accumulation, raising the hypothesis that certain toxic substances impair anti-oxidant systems and increase oxidative stress and reactive oxygen species (ROS) production. Among these, several tryptophan-derived metabolites have been identified as ROS-generating uraemic toxins, including indoxyl sulphate and kynurenines (3-hydroxykynurenine, kynurenic acid and hydroxyanthranilic acid), 141 which we discuss further (Figure 4).

The role of CKD-associated pathological changes in the increased risk of atherosclerosis and other cardiovascular diseases. CKD-specific primary pathologic changes (such as uraemic toxins, albuminuria, anaemia and others) (depicted in fuchsia box) lead to secondary damage to cardiovascular system (such as endothelial dysfunction, oxidative stress and others) (depicted in magenta box) which lead to the remodelling of the myocardium and blood vessels (depicted in red box). These processes further contribute to the development and progression of atherosclerosis and other cardiovascular diseases.
Indoxyl sulphate has been shown to contribute to vascular injury by increasing endothelial susceptibility to oxidative stress. Experiments on human umbilical vein endothelial cells (HUVEC) demonstrated that indoxyl sulphate reduces cell viability, increases ROS production and causes mitochondrial dysfunction – manifested as reduced mitochondrial membrane potential, DNA copy number and mass. 142 Additionally, indoxyl sulphate up-regulates miR-34a, which targets neurogenic locus notch homolog protein 1 (Notch1), affecting its downstream pathways and impairing endothelial cell proliferation and migration while promoting apoptosis. 143
Further studies have shown that NADPH oxidase-derived ROS plays a crucial role in activating the MAPK/NFκB/Activator protein 1 (AP-1) pathway in indoxyl sulphate-treated HUVEC. This pathway activation enhances the production of E-selectin, a key adhesion molecule responsible for recruiting leukocytes to inflammatory sites. 144 In human aortic endothelial cells, indoxyl sulphate treatment increased NADPH oxidase activity while reducing endothelial nitric oxide synthase (eNOS) activity and nitric oxide (NO) production, thereby promoting endothelial dysfunction. 145 Similar results were observed in another study, where indoxyl sulphate-treated HUVEC exhibited increased ROS production alongside decreased eNOS and VE-cadherin expression. 146 In vivo, CKD mice acutely and chronically exposed to indoxyl sulphate showed elevated expression of intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1), indicating endothelial activation and atherosclerosis progression. 147
Toxic metabolites from the tryptophan–kynurenine pathway have also been implicated in endothelial dysfunction, inflammation and atherosclerosis.148 –150 For example, indoleamine 2,3-dioxygenase 1 (IDO1), the enzyme responsible for the first and rate-limiting step in tryptophan metabolism along this pathway, is up-regulated in coronary atherosclerotic plaques from patients with unstable angina pectoris compared to stable cases. IFNγ and TNFα further induce IDO1 expression, increasing the kynurenine/tryptophan ratio and NFκB activity in macrophages. Inhibition of the kynurenine-binding aryl hydrocarbon receptor by Epacadostat reduces kynurenine-induced tissue factor expression in activated macrophages, suggesting that enhanced IDO1 expression contributes to atherosclerosis progression through increased oxidative stress and inflammation. 151
However, some kynurenine metabolites exhibit anti-atherosclerotic effects. For example, kynurenic acid enhances peroxisome proliferator-activated receptor delta (PPARδ) expression, reduces NFκB phosphorylation and decreases pro-inflammatory cytokine release in LPS-treated HUVEC and macrophages. Kynurenic acid also mitigates LPS-induced inflammation and apoptosis while promoting haem oxygenase-1 (HO-1) expression, which suppresses inflammation in HUVEC. 152 Thus, the balance between pro-atherogenic and anti-atherogenic tryptophan metabolites may play a pivotal role in the development and progression of atherosclerosis.
There is a bidirectional relationship between atherosclerosis and CKD. Kidney dysfunction exacerbates atherosclerosis through the accumulation of uraemic toxins, increased oxidative stress, and vascular damage, while atherosclerotic plaque rupture contributes to kidney ischaemia and further renal injury. These interactions highlight the importance of managing both conditions together to reduce cardiovascular risk and prevent further organ damage. Early intervention and monitoring of kidney function in patients with atherosclerosis are essential for improving patient outcomes.
Kidney stones
KS are a common multifactorial urological condition characterised by the formation, retention, deposition and occasional passage of crystal aggregates in the urinary tract. Factors such as diet, sex, age and race influence the type of stones formed and their recurrence rate, with obesity and metabolic syndrome being major risk factors for KS development. Globally, calcium oxalate is recognised as the dominant component of KS.153 –155 KS is currently recognised as a risk factor for CKD, 156 diabetes, 157 CVD, 158 and bone fractures. 159 Below, we review recent findings on the relationship between KS and atherosclerosis.
The role of atherosclerosis in KS development
The vascular theory of Randall plaque formation posits a connection between atherosclerosis-like processes and calcium oxalate KS development, suggesting that the repair of injured papillary vasculature leads to calcification near vessel walls, which subsequently transform into KS. Renal physiological properties, including turbulent blood flow at the papillary tip, higher osmolality between the renal cortex and papilla, and a decreasing oxygen-carrying capacity gradient, may facilitate atherosclerotic-like inflammatory responses with perivascular calcification, contributing to Randall plaque formation and KS progression. 160 A long-term observational study in young individuals in the United States found an association between urinary stone formation and subclinical carotid atherosclerosis, suggesting shared pathophysiological mechanisms. 161
Elevated serum triglyceride (TG) levels have been specifically linked to an increased risk of urinary stones, whereas no significant associations were observed with other circulating lipids (e.g. LDL-C, HDL-C, APOA, APOB) or lipid-lowering treatments (HMGCR and PCSK9 inhibitors). 162 However, another study found strong associations between carotid intima-media thickness (IMT), carotid scores, elevated serum TC and LDL levels and the presence of both calcium oxalate (CaOx) and calcium phosphate (CaP) stones, connecting dyslipidaemia, carotid atherosclerosis and KS. 163
A high-fat diet is a well-established risk factor for KS. Animal studies have demonstrated that such diets contribute to acidic urinary pH, promote the formation of uric acid and calcium-containing crystals and lead to renal injury and crystal retention in the urothelium (Figure 5). 164 Similarly, experiments on rats with metabolic syndrome showed that hyperoxaluria exacerbates renal CaOx crystal retention, causes severe morphological changes and significantly impairs renal function compared to control rats without metabolic syndrome.165,166

Mechanism of kidney stone formation. Unbalance diet (such as oxalate-rich diet) or genetic factors caused hyperoxaluria, hypercalciuria, hypocitraturia, hyperuricosuria or hyperuricosuria. Subsequently, these factors lead to the supersaturation of salts such as CaOx or uric acid or CaP, their nucleation, growth, aggregation and retention in the arterial wall, which resulted in kidney stone formation. Surplus ROS is formed by high uric acid or high oxalate, caused renal injury and promoted crystal nucleation and aggregation, thus facilitating kidney stones formation.
Oxidised LDL (oxLDL), a major driver of atherosclerosis, has been proposed as a causative link between atherosclerosis and KS. Although most oxLDL is taken up by the liver, approximately 2% is absorbed by the kidneys. 167 Under pathological conditions, renal uptake of oxLDL may increase. IL-1β has been shown to up-regulate the expression of the lectin-like oxLDL receptor 1 (LOX-1) in glomerular mesangial cells, promoting oxLDL uptake and accumulation, which can lead to lipid nephrotoxicity and podocyte damage.168,169 OxLDL also enhances ADAM10 and CXCL16 expression, stimulating podocyte migration. 170 Interestingly, while no difference in circulating oxLDL levels was observed between patients with and without KS, KS patients exhibited higher urine oxLDL levels, which correlated with serum CRP levels and stone size. Additionally, increased renal oxLDL uptake was observed in KS patients, but not in controls without KS. 171
The application of statins, commonly used for cholesterol lowering, has also shown benefits in KS management, further highlighting the close link between atherosclerosis and KS. Statins stabilise lipid parameters and provide protection against KS. 172 In hyperlipidaemic and hyperoxaluric rat models, atorvastatin reduced renal CaOx stone deposition by decreasing free radical production and increasing osteopontin (OPN) expression, a key anti-lithic protein. Interestingly, this reduction in renal calcium deposition was accompanied by increased urinary bladder calcium crystals, suggesting a shift in the dominant crystal composition from CaOx to calcium phosphate. 173
In another rat model, atorvastatin increased superoxide dismutase (SOD) activity, mitigated kidney damage, reduced ER stress, apoptosis, and autophagy and decreased crystal deposition. Conversely, inhibiting SOD with diethyldithiocarbamic acid (DETC) aggravated crystal deposition and renal pathology, indicating the central role of SOD in atorvastatin’s protective effects. 174 Other studies confirmed that atorvastatin alleviates CaOx crystal-induced renal damage by suppressing oxidative stress and inflammation. Mechanistically, atorvastatin increased SOD levels, reduced malondialdehyde (MDA), lactate dehydrogenase (LDH) and ROS levels and inhibited the activation of TLR4/NFκB and NLRP3 inflammasome pathways, thereby decreasing pro-inflammatory cytokine secretion (IL-1β, IL-6, IL-18 and TNFα). 175 These findings suggest that atorvastatin’s antioxidant and anti-inflammatory properties may offer a novel therapeutic approach for KS prevention and treatment.
KS leads to atherosclerosis
A growing body of evidence demonstrates a strong association between KS and an elevated risk of ASCVD. Meta-analyses of cohort studies have shown that patients with KS are at increased risk of CHD and stroke.176 –178 Stratified analyses across racial and ethnic groups revealed that non-Hispanic Black individuals with KS were 2.24 times more likely to experience an ASCVD event within the next decade compared to controls without KS. 179 Similarly, the Taiwan patients with KS had higher risk of MI, stroke and total cardiovascular events in the future.180,181 In these populations, patients with CaOx and CaP KS were found to exhibit elevated serum TC and LDL levels, coupled with lower urinary citrate and increased carotid IMT, linking KS, dyslipidaemia and atherosclerosis. 163 Korean studies have further highlighted the association of KS with heightened risks of stroke and ischaemic heart disease. 182 Key pathogenic factors linking KS and ASCVD include endothelial dysfunction, hyperuricaemia and systemic inflammation.183 –185
Experimental studies using mouse models of CaOx KS have identified significant upregulation of genes involved in atherosclerosis, bone metabolism and calcium homeostasis. A range of atherosclerosis-related genes showed marked upregulation (more than tenfold), including adhesion molecules (CD44, VCAM-1), extracellular matrix components (matrix metallopeptidase 3, plasminogen activator inhibitor-1, collagen type III alpha 1 chain, fibrinogen beta chain, leukaemia inhibitory factor and macrophage scavenger receptor 1) and inflammatory mediators (chemokine ligand 2, C-C chemokine receptor type 1, chemokine ligand 1, secreted phosphoprotein 1 and IL-6). 186 These findings provide molecular insights into the mechanisms linking renal CaOx deposition with atherosclerosis.
The association between atherosclerosis and KS disease is driven by common mechanisms such as inflammation, dyslipidaemia and perivascular calcification. Atherosclerosis-like responses in kidney tissue promote stone formation, while KSs aggravate vascular inflammation and increase the risk of ASCVD. The up-regulation of atherosclerosis-promoting genes in KS disease further underscores the need for a holistic approach to treating both conditions. Effective management of lipid metabolism and inflammation could help reduce the risk of both cardiovascular and KS disease.
Thyroid dysfunction
Subclinical thyroid dysfunction, encompassing subclinical hypothyroidism (SHypo) and subclinical hyperthyroidism (SHyper), is characterised by abnormal serum thyroid-stimulating hormone (TSH) levels, while levels of free thyroid hormones (thyroxine [T4] and triiodothyronine [T3]) remain within the normal range. SHypo is defined by elevated TSH levels, while SHyper is marked by TSH levels below normal. SHypo is more prevalent (3%–10%) compared to SHyper (0.7%–10%). 187 This section explores the associations between SHypo/SHyper and increased ASCVD risk, biomarkers and underlying molecular mechanisms.
SHypo/SHyper and ASCVD risk
SHypo can be classified as mild or severe based on TSH levels, with most cases associated with antithyroid peroxidase antibodies, characteristic of Hashimoto’s thyroiditis. 188 SHypo is more common in iodine-sufficient regions, where excessive iodine intake may exacerbate its incidence. 189 Increasing evidence suggests that SHypo contributes to ASCVD risk through dyslipidaemia, diastolic left ventricular dysfunction, hypertension, insulin resistance and direct cardiovascular effects mediated by elevated TSH.190,191
For example, a prospective cohort study in Korean adults with high CVD risk found that SHypo was associated with increased all-cause mortality and CVD events, particularly in individuals aged < 65 years. 192 This finding aligns with meta-analyses showing that SHypo is linked to higher CVD event rates and all-cause mortality, though the association was less pronounced in low-risk populations. 193 Another meta-analysis, however, observed a connection between SHypo and CHD mortality but not other cardiovascular endpoints, such as heart failure or atrial fibrillation, with stronger associations in participants aged < 65 years. 194 Additionally, decreased central sensitivity to thyroid hormone in SHypo has been linked to hyperuricaemia and increased CVD risk. 195
Studies in children with SHypo have also highlighted early markers of atherosclerosis, including increased epicardial fat thickness (EFT) and CRP levels, along with reduced brachial artery flow-mediated dilation (FMD).196,197 While adult patients with SHypo did not show significant differences in EFT, they exhibited increased carotid IMT and reduced aortic velocity propagation compared to controls. 198
Even within normal thyroid hormone ranges, higher TSH levels have been associated with adverse cardiometabolic profiles, including elevated glucose, haemoglobin A1c and TGs. 199 Dyslipidaemia (elevated LDL, TC, TG and reduced HDL), insulin resistance, CRP and homocysteine levels further link SHypo to ASCVD risk.200 –204 Conversely, some studies have failed to demonstrate a significant correlation between SHypo or rising TSH and the 10-year risk of adverse cardiac events. 205 These discrepancies may reflect the need for more nuanced analyses that account for ethnicity, genetics, age and comorbidities.
SHyper, defined by low serum TSH levels, is more prevalent in iodine-deficient regions and categorised into Grade 1 (low but detectable TSH) and Grade 2 (undetectable TSH). 206 Like SHypo, SHyper has been linked to ASCVD and arrhythmias.207 –209
Meta-analyses of prospective studies show that SHyper is associated with increased risks of CHD, CHD mortality and total mortality.194,210 SHyper has also been linked to a higher risk of atrial fibrillation, 211 In patients with acute coronary syndrome (ACS), low triiodothyronine syndrome (low T3) was associated with increased all-cause and cardiac mortality, whereas SHypo or SHyper showed no significant effect. 212
In conclusion, further studies are warranted to clarify the interplay between thyroid dysfunction, genetic predispositions, lifestyle factors and ASCVD risk and to identify effective screening and treatment strategies.
Mechanisms underlying thyroid dysfunction and increased ASCVD risk
Dyslipidaemia, hypertension, endothelial dysfunction and a hypercoagulable state have been proposed as the major risk factors linking thyroid dysfunction to ASCVD. 213 This connection may be explained through several functional properties of thyroid hormones (Figure 6). First, thyroid hormones promote the expression of hepatic LDL receptor, which regulates cholesterol transport and lipid metabolism by mediating the hepatic uptake of LDL from plasma. 214 Second, thyroid hormones play a crucial role in regulating BP and heart rate.215,216 Third, thyroid hormones affect other risk factors, such as hypercoagulability and hyperhomocysteinaemia, which can facilitate atherosclerosis development in patients with thyroid dysfunction. 217 Recent studies have explored the contribution of thyroid dysfunction to atherosclerosis development.
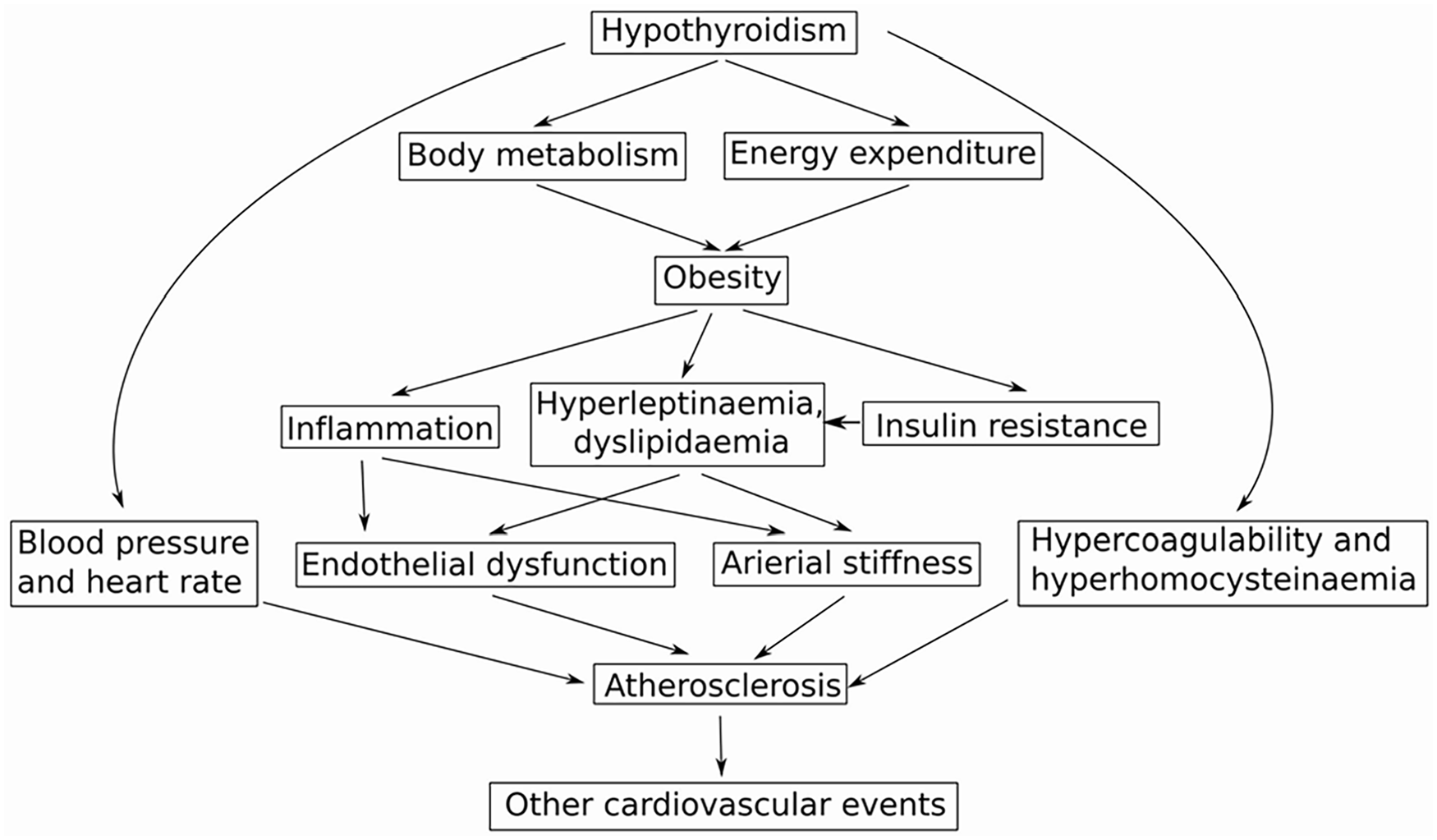
The summary of the pathogenic mechanisms connecting the increased risk of atherosclerosis development in hypothyroidism. Thyroid hormones regulate food intake, metabolism of lipids and glucose, thus affecting energy homeostasis and body weight. Increased adiposity caused hyperleptinaemia, which stimulated TSH secretion, which, in turn, promoted a differentiation of pre-adipocytes into adipocytes, thus closing the vicious cycle. Obesity is often accompanied by insulin resistance, which can stimulate leptin release and lead to hyperleptinaemia. The increased production of inflammatory cytokines in obesity reduced iodide uptake and may induce thyroid gland malfunction. Additionally, the described processes negatively affected blood vessels, causing endothelial dysfunction and arterial stiffness, and thus facilitating development of atherosclerosis and other cardiovascular diseases. The direct effects of thyroid hormones on heart rate, blood pressure, homocysteine levels and coagulation system are another processes increasing risk of ASCVD.
The reduction in brachial artery FMD is known as the primary sign of endothelial dysfunction and atherosclerotic changes. Thus, SHypo patients had lower FMD compared to euthyroid controls. However, treatment with levothyroxine, a synthetic form of the thyroid hormone thyroxine, which has been the primary treatment for SHypo since 1927, 218 normalised FMD in SHypo patients.219,220
Adhesion molecules have also been associated with hyperthyroidism. SHyper patients exhibited higher VCAM-1 levels and a lower ankle-brachial index (ABI) – a simple test for peripheral artery disease – compared to euthyroid subjects. However, treatment with antithyroid drugs (thioamides, which block the biosynthesis of thyroid hormones) decreased free T4 levels, increased ABI and reduced VCAM-1 levels. 221 In a similar study, treatment of SHyper patients with thioamides (propylthiouracil and methimazole) normalised thyroid hormone levels to euthyroid conditions and was accompanied by a reduction in adhesion molecule levels (ICAM-1, VCAM-1 and E-selectin). 222
Moreover, the molecular mechanism underlying the association between thyroid hormones and inflammation was elucidated in experiments on TSH receptor (Tshr)-deficient ApoE−/− mice fed a Western diet. The ablation of the TSH receptor in ApoE−/− mice reduced plaque area, macrophage burden in the plaques and the expression of pro-inflammatory cytokines (IL-1β, IL-6, TNF-α and CCL2) in the aorta, alongside a decrease in serum levels of IL-6 and TNFα, thus suppressing vascular inflammation and atherosclerosis progression. Further in vitro experiments on macrophages treated with TSH revealed a direct up-regulatory effect on inflammation marker expression (e.g. NOS2, IL-6, CX3CL1 and TNF-α) while down-regulating markers associated with inflammation resolution (e.g. ARG1, PPARγ, LXRA and ABCA1). TSH treatment also activated MAPKs (ERK1/2, p38α and JNK) and the IκB/p65 pathways in macrophages, promoting the production of pro-inflammatory cytokines and monocyte recruitment. 223 Further research has shown that TSH activated Toll-like receptor 4 (TLR4), 224 which is known to play a role in activating innate and adaptive immunity, inflammation, anti-tumour activity and other processes.225,226 Subsequent application of specific inhibitors and siRNA demonstrated that the downstream pro-inflammatory signalling of the TSH receptor is mediated through G proteins: G13 (for ERK/p38) and G15 (for phospholipase C (PLC)/protein kinase C (PKC)/IκB pathways). 227 In total, these findings highlight the detailed molecular mechanisms by which TSH aggravates vascular inflammation and promotes atherosclerosis, providing new insights into the treatment and prevention of atherosclerosis through monitoring thyroid hormone levels as an independent risk factor.
Thyroid dysfunction, including both hypothyroidism and hyperthyroidism, is closely linked to increased ASCVD risk. Thyroid hormones influence lipid metabolism, endothelial function and coagulation, all of which play crucial roles in the development of atherosclerosis. Mechanisms involving TSH receptor activation, MAPK pathways and inflammation contribute to vascular dysfunction and atherosclerotic progression. Understanding these molecular mechanisms offers new insights into managing thyroid dysfunction as an independent risk factor for ASCVD and atherosclerosis.
Conclusion
Atherosclerosis has long been known to be associated with the development of various multiorgan pathologies characterised by chronic inflammation, oxidative stress and dyslipidaemia. However, the significant advances made over the past decade have greatly expanded our understanding of how atherosclerosis-associated pathological changes affect the metabolism of vascular cells in different tissues and organs. In this review, we focused on the association between atherosclerosis and stroke, Alzheimer’s disease, NAFPD, CKD, KS disease and thyroid dysfunction. It is challenging to distinguish the specific pathways affected by atherosclerosis, as many of the adverse effects associated with atherosclerosis are also attributed to the manifestation of other closely related conditions (such as metabolic syndrome, diabetes mellitus, obesity and others) that share common risk factors with atherosclerosis (primarily hypertension, dyslipidaemia, smoking, advanced age, stress, genetic factors and many others).
A strong association has been established between atherosclerosis and IS, with napkin-ring sign plaques, a ‘spotty’ pattern of plaque calcification, and elevated serum levels of aldosterone, CRP, and ELAVL1 protein being potent stroke biomarkers. Interestingly, atherosclerosis and Alzheimer’s disease have been shown to promote each other through several pathways. Notably, the well-studied C/EBPβ/AEP signalling pathway has been demonstrated to connect atherosclerosis and AD through ApoE-mediated vascular dysfunction. Additionally, the ε4 allele of the ApoE gene has been associated with more severe forms of atherosclerosis and a higher rate of cognitive decline in AD.
Furthermore, CKD and atherosclerosis have been shown to exacerbate one another. Kidney dysfunction increases the accumulation of certain uraemic toxins, which impair the antioxidant system, increase ROS generation and promote oxidative damage, thereby exacerbating vascular dysfunction and the development of atherosclerosis. On the other hand, the rupture of atherosclerotic plaques can release CC into the bloodstream, which can become lodged in arterioles, leading to ischaemia and infarction in various tissues and organs, including the kidneys. Similarly, atherosclerosis and KSs have been linked through dyslipidaemia and oxLDL accumulation. Atherosclerosis-like responses to inflammation and perivascular calcification have been shown to promote KS formation. KSs, in turn, up-regulate a wide range of atherosclerosis-promoting genes (such as adhesion molecules, extracellular matrix molecules and pro-inflammatory cytokines), which increase the risk of ASCVD.
The role of atherosclerosis in pancreas dysfunction has been mechanistically explained by atherosclerosis-mediated reductions in blood flow to the pancreas, which causes islet hypoxia and β-cell dysfunction, leading to NAFPD and possibly accompanied by NAFLD and diabetes. Finally, dyslipidaemia, hypertension, endothelial dysfunction and a hypercoagulable state have been proposed as the major risk factors linking thyroid dysfunction and ASCVD. In vivo and in vitro experiments have demonstrated that thyroid hormones directly activate the expression and production of pro-inflammatory cytokines and adhesion molecules. In particular, TSH has been shown to aggravate vascular inflammation and promote atherosclerosis development by activating MAPKs, IκB/p65, TLR4 and PLC/PKC signalling pathways, at least partially through G proteins (G13 and G15).
The results discussed suggest that regular monitoring and timely treatment of atherosclerosis-related vascular risk factors may be a valuable strategy for treating and preventing Alzheimer’s disease, pancreas and thyroid dysfunctions, KSs and CKD. On the other hand, the pathologies of many organs may manifest through ASCVD, complicating diagnosis and treatment and potentially leading to life-threatening conditions. Overall, further studies deciphering the diverse mechanisms by which atherosclerosis is associated with multiple organ pathologies would help generate new therapeutic strategies to mitigate the adverse effects of atherogenesis on other organs.
Footnotes
Acknowledgements
Not applicable.
Author contributions
S.A.D and A.N.O. conceptualised the manuscript; S.A.D. writing – original draft preparation; A.V.C., N.V.E., A.L.R., A.E.K., V.N.S. and A.N.O. review and editing; A.L.R. and N.V.E. validation; A.V.C. and A.E.K. formal analysis; V.N.S and A.N.O. obtained funding and supervised. All authors have read and agreed to the published version of the manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship and/or publication of this article: This work was supported by Russian Science Foundation, Grant# 24-15-00123 (conceptualisation; writing – original draft preparation, writing – review and editing; formal analysis; validation; funding acquisition; project administration).
Ethical considerations
Not applicable.
Informed consent
Not applicable.
Trial registration
Not applicable.