
Abstract
Objectives
The aim of this study was to report the spectrum of genetic variations and clinical phenotype in a Vietnamese cohort with confirmed Brugada syndrome (BrS) using the whole exome sequencing (WES).
Methods
Fifty patients with confirmed BrS were included in this study. Genomic DNA samples were extracted from peripheral blood and conducted for WES. The variants were annotated using ANNOVAR. The variants in the 13 reported genes associated with BrS were filtered, predicted the functional impact using eight computational tools, and classified according to the 2015 ACMG guidelines.
Results
Arrhythmic events were documented in one-fifth of the participants. Twenty-four probands were identified to carry 36 variants in 13 genes. Majority of the variants in our study was SCN5A variants (9/36 variants, 25%), followed by KCNH2 variants (5/36 variants, 14%). The prevalence of SCN5A carriers was 16%; while the prevalence of minor gene carriers was less than 10%. Nine novel missense variants were identified, including four missense SCN5A variants (p.E901D, p.F853L, p.L377F, and p.H184R), two missense ANK2 variants (p.S2845L and V1497L), one missense CACNA1C variant (M1126V), one missense DSP variant (p.K478N), and one intron splicing JUP variant (c.1498-5G>C).
Conclusion
Our study underscores the primary significance of the SCN5A gene in BrS, as indicated by variant prevalence, carrier rates, pathogenicity per ACMG classification, in silico predictions, and its correlation with clinical phenotypes. Longitudinal study with larger sample size, pedigree, Sanger sequence confirmation, and functional analysis is recommended.
Introduction
Brugada syndrome (BrS), first described in 1992, is a hereditary condition of cardiac electrical conduction, which associated with the risk of sudden cardiac death (SCD) secondary to polymorphic ventricular tachycardia and ventricular fibrillation.1,2 BrS is diagnosed by the presence of coved-type ST-segment elevation in the right precordial leads (V1–V3) on surface electrocardiography (ECG) spontaneously or after induced by IC arrhythmias drugs, also called a type 1 Brugada ECG pattern, in the absence of structural cardiac disease. 1 Since the majority of BrS patients are asymptomatic, the condition is usually accidentally discovered during a regular evaluation or a family screening when an ECG shows abnormalities. 3 Some patients exhibit a range of symptoms, such as syncope (30%), paroxysmal nocturnal dyspnea (12%), ventricular tachycardia/fibrillation (6%), and SCD (6%). SCD is often the first manifestation of BrS, predominantly in adult males at night or during rest.4,5 At least 4% of SCDs are attributed to BrS, with at least 20% of those occurring in otherwise healthy individuals. 3 Patients with a history of cardiac syncope and those who have had an attempted cardiac arrest or recorded spontaneous prolonged ventricular tachycardia (VT) are at the highest risk for SCD. Moreover, asymptomatic patients have a yearly chance of 0.5–1.5% of developing potentially fatal arrhythmias. 4 This highlights the significance of determining the genetic basis of BrS in order to identify asymptomatic genetic carriers who are at risk for developing SCD.
Over 500 variants in 43 genes are currently considered to account for more than 35% of BrS cases, most notably those involved in regulating sodium current (Ina), L-type calcium channels (Ica), and transient outward potassium channels (Ito). 6 However, most of these genes are associated with limited or disputed evidence, and SCN5A remains the only gene with a well-established clinical actionability for BrS. It is estimated that 25–30% of all confirmed cases are due to definitive pathogenic variants in the SCN5A gene, while all other genes together are only responsible for up to 10% of the diagnosed cases, and their association with BrS is not conclusively proven.7,8 These genes are categorized as minor genes with insufficient BrS-related evidence.9,10 As of now, there is a lack of published data and evidence on genetic background and their impact on the phenotypic characteristics of Vietnamese BrS population. A study on 117 Vietnamese BrS patients identified this gap by primarily focusing on evaluating variants in the SCN5A gene, however, evidence on the presence and role of minor genes remains unaddressed. 11
To clarify the genetic association with BrS, we aimed to report the spectrum of genetic variants and clinical phenotype in a Vietnamese cohort with confirmed BrS using the whole exome sequencing (WES) technique.
Materials and methods
Study population
A total of 50 unrelated patients with spontaneous type 1 BrS were included from Vietnam National Heart Institute and Hanoi Medical University Hospital from March to December 2023. A definitive diagnosis of BrS was made using the 2015 updated criteria provided by the European Society of Cardiology, with the presence of a spontaneous type 1 Brugada ECG pattern (an ascending and high take-off of ≥2 mm at the end of the QRS duration in ≥1 right precordial leads (V1–V3, which are placed in the second, third, or fourth intercostal space), followed by a coved or rectilinear downsloping ST-segment and a negative symmetric T wave). Patients with an acquired cause of a type 1 ECG pattern and/or structural cardiac disease were excluded.
Clinical features
The following clinical features were obtained: age, gender, and family history of SCD/BrS. Arrhythmic events included syncope, sudden cardiac arrest (SCA), and/or a documented ventricular tachycardia/ventricular fibrillation (VT/VF). If the patient had not experienced any arrhythmic events and had exhibited a Brugada type 1 electrocardiogram, a programmed ventricular stimulation was conducted to assess whether the patient could induce VT/VF. It is important to note that, in this study, the initiation of ventricular arrhythmias during programmed ventricular stimulation was not considered an arrhythmic event. If the patient had a history of prior implantation of an implantable cardioverter-defibrillator (ICD), the information regarding the timing and reason for the device implantation, ventricular arrhythmias documented on the ICD, and the number of appropriate shocks recorded on the ICD was recorded. All the information had been recorded in detail in patient's medical charts.
A qualified healthcare worker conducted 12-lead ECG recordings at a speed of 25 mm/s during the administration. The task of analyzing the electrocardiograms was assigned to a board-certified cardiologist who remained blind to clinical and genetic data. Heart rate, QRS axis, durations of P wave, and QRS intervals were measured in accordance with established criteria. 12 The QT interval was corrected for the heart rate according to Bazett's formula (QTc = QT/√RR), while the heart rate-corrected JT interval (JTc) was calculated by the following formula: JTc = QTc − QRS. 13 Bundle branch block and atrioventricular block was defined according to the standard AHA criteria, as previously reported. 14 Early repolarization was defined as the presence of J waves or J-point elevation followed by a horizontal or downsloping ST segment in inferior leads or lateral leads. 15 Fragmented QRS was defined as the presence of two or more notches in the QRS complex on leads from V1 to V3. 16 The aVR sign was present when the R/Q ratio is ≥0.75 or the R wave is ≥0.3 mV in the aVR lead. 17 The Tpeak–Tend interval was calculated from the Tpeak position, which was the peak/bottom of the T wave, to the Tend position, which is the intersection of the isoelectric line with the tangent to the downslope of the T wave. The Tpeak–Tend interval was the result of averaging measurements from three consecutive beats across all leads from V1 to V6. 18
Whole exome sequencing
Peripheral blood was obtained from the patients for WES analysis. The genomic DNA samples were extracted from the peripheral blood using a standard DNA extraction protocol. The extracted genomic DNA was fragmented into fragments of 150–200 bp in length, followed by library preparation using established Illumina paired-end procedures. The adaptor-ligated libraries were amplified through PCR, and individual library components were used to generate an equimolar pool. The Agilent SureSelectXT Target Enrichment System was then used to amplify each pool, enriching the targets to be sequenced (Agilent Technologies Inc., Santa Clara, CA, USA). The target exon and adjacent splicing areas were captured and enriched by Roche KAPA HyperExome (Roche Molecular Systems, Inc.) The capture kit was able to capture the target areas with the average sequencing depth greater than 100× and 90% of target areas with a minimal coverage of 20×. The exome-enriched libraries were sequenced on the Illumina HiSeq 2000 platform (Illumina, San Diego, CA, USA) in accordance with the manufacturer's protocols, generating paired-end sequencing reads of 100 bp in length. Each sample was sequentially ordered for each lane.19,20
Variant calling and annotation
Upon completion of WES, the quality of the raw reads was assessed, and low-quality reads were removed, along with trimming of 3′/5′ adapters, using the Trim Galore tool (version 0.4.4) for quality control. The clean reads were mapped to the human reference genome (University of California Santa Cruz, UCSC build hg19) using the Burrows–Wheeler Aligner (BWA, version: 0.7.17-r1188). The quality scores were recalculated and the reads were realigned to the reference genome using the Genome Analysis Toolkit (GATK, version: 3.5-0-g36282e4). Following the elimination of duplicate reads, insertions–deletions (InDels) and single nucleotide polymorphisms (SNPs) were identified using the GATK or Sequence Alignment/Map (Samtools, version: 1.3.1) programs. The variants were annotated using ANNOVAR.
Variant identification and pathogenic risk classification
In this study, we focused on variants of reported genes associated with BrS, as identified in a prior systematic review. These genes include ABCC9, KCNE2, SCN2B, ACTC1, KCNE3, SCN3B, AKAP9, KCNE5, KCNE1L, SCN4B, ANK2, KCNH2, SCN5A, CACNA1C, KCNJ2, SCN10A, CACNA2D1, KCNJ5, SEMA3A, CACNB2, KCNJ8, SNTA1, CASQ2, KCNQ1, TMEM43, CAV3, RANGFR, MOG1, TNNI3, DSC2, MYBPC3, TNNT2, DSG2, MYH7, TPM1, DSP, MYL2, TRPM4, FLNC, MYL3, LMNA, GPD1L, PKP2, PLN, HCN4, CBL, JUP, NOS1AP, tRNA-Ala, KCND3, RYR2, tRNA-Gln, KCNE1, SCN1B, and tRNA-Met. 21
To predict the functional impact of the variants, we employed eight computational tools, comprising six individual predictors: Mutation Assessor (http://mutationassessor.org/r3/), Polyphen-2 (http://genetics.bwh.harvard.edu/pph2/), Mutation Taster (https://www.mutationtaster.org/), PROVEAN (http://provean.jcvi.org/index.php), FATHMM (http://fathmm.biocompute.org.uk/), SIFT (https://sift.bii.a-star.edu.sg/index.html), and two meta-scores: BayesDel addAF and MetaSVM. These algorithms were assessed based on the structural and functional aspects of the targeted protein and its evolutionary conservation in the sequence. In the presence of more than half of the in silico tools predicting pathogenic/benign outcomes, the variant was classified as predicted pathogenic/benign; otherwise, the variant will be categorized as having uncertain pathogenicity.
In this study, genetic variants were interpreted using the position statement from the European Society of Cardiology Council on Cardiovascular Genomics in 2022. 22 Evidences interpreted from each variant was fitted on a scaled point system described from a previous study. 23 Each rule triggered is assigned a specific number of points according to the evidence strength: Supporting (1 point), Moderate (2 points), Strong (4 points), and Very Strong (8 points). The total score is calculated by summing the points from pathogenic rules and subtracting the points from benign rules. The final classification is determined by comparing the total score to the following thresholds: pathogenic if greater than or equal to 10, likely pathogenic if between 6 and 9 inclusive, uncertain significance if between 0 and 5, likely benign if between −6 and −1, and benign if less than or equal to −7.
Novel variants referred to those variants that had not been reported in databases including ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), LOVD (https://www.lovd.nl/), dbSNP (https://www.ncbi.nlm.nih.gov/snp/), and the Genome Aggregation Database (gnomAD) (https://gnomad.broadinstitute.org/).
Statistical analysis
We described qualitative variables as frequencies and percentages and quantitative variables as means (standard deviation, SD) or medians (interquartile range, IQR). Continuous variables with a normal distribution were compared using a t-test or one-way ANOVA, whereas continuous variables with an abnormal distribution were compared using the Wilcoxon rank-sum test or Kruskal–Wallis test. To compare categorical variables, the chi-square test or Fisher exact test was employed. The Kaplan–Meier method was employed to estimate survival endpoints, with survival time defined as the interval from birth to the occurrence of the first arrhythmic events, including syncope, SCA, and documented VT/VF, whichever came first. In cases where patients were included in the study without experiencing any arrhythmic events, the admission date would be considered as the time of administrative censoring. The log-rank test was utilized to compare survival distributions across groups (non-carriers vs SCN5A carriers vs minor gene carriers). A p-value of less than 0.05 was considered statistically significant. R language version 4.3.2 was used for all analyses.
Ethical consideration
The study was conducted in accordance with the Declaration of Helsinki and approved by the Institutional Review Board of Hanoi Medical University under decision No. 682/GCN-HĐĐĐNCYSH-ĐHYHN dated 16 February 2023. Written informed consent was obtained from the patients before participating in the study. The investigators were responsible for protecting the privacy and confidentiality of patients as per Vietnam's regulations and Good Clinical Practice.
Results
A total of 50 unrelated BrS patients were included in this study, of which 96% were male and the mean age at diagnosis was 47 ± 12. Reported family history of SCD and BrS was 13% and 3.1%, respectively. Arrhythmic events were documented in one-fifth of the participants. Electrophysiological study (EPS) was conducted in 70% of participants, with successful induction of VT/VF observed in 23% of cases. The prevalence of VT/VF induced was higher in the SCN5A carriers compared to the wild-type group (67% vs 13%, p = 0.018), meanwhile, this prevalence tended to be higher in the minor gene carriers compared to the wild-type group (44% vs 13%, p = 0.076). Trends toward higher prevalence of aVR signs, type 1 BrS pattern in peripheral leads, and higher S wave amplitude in D2 were also observed in the SCN5A carriers compared to the wild-type group, while no other differences were noted between the minor gene carriers and the wild-type group (Table 1). Kaplan–Meier analysis did not reveal a significant difference in median time to ventricular arrhythmia events between SCN5A carriers and wild type, as well as between minor gene carriers and wild type (Figure 1).

Kaplan–Meier estimates of time to arrhythmic events among the study participants, stratified by variant type (n = 50).
Participant characteristics (n = 50).

Variables in bold were statistically significant.
SCN5A: sodium voltage-gated channel alpha subunit 5; SCD: sudden cardiac death; BrS: Brugada syndrome; EPS: electrophysiological study; VT: ventricular tachycardia; VF: ventricular fibrillation; bpm: beat per minute; AVB: atrioventricular block.
Four patients were multivariant carriers, harboring variants in both SCN5A and minor genes.
Using WES technique, 36 variants in 13 genes associated with BrS were identified in 24 patients (48%), including SCN5A, KCNH2, DSP, ANK2, JUP, MYBPC3, CACNA1C, DSC2, FLNC, HCN4, KCNE2, PKP2, and SCN3B (Table 2). Among these, SCN5A had the highest number of variants (nine variants, 25%); followed by KCNH2 (five variants, 14%); DSP (four variants, 11%). The remaining genes had variant rates < 10%. In this study, the prevalence of SCN5A carriers was 16%, whereas the prevalence of carriers of minor genes was less than 10%.
The variants identified in BrS probands.

dbSNP: database for single nucleotide polymorphisms; ExAC: exome aggregation consortium; ACMG: American College of Medical Genetics and Genomics; Pt: patient; P: pathogenic; U: uncertain; B: benign; BP: evidence of benign influence supporting; BS: evidence of benign-impact strong; LP: likely pathogenic; NA: not available; PM: evidence of pathogenicity moderate; PP: evidence of pathogenicity supporting; PS: evidence of pathogenicity strong; VUS: variant of uncertain significance.
The gnomAD % was extracted from gnomAD version 4.1 data, specifically referring to the Allele Frequency of the East Asian population.
Variants in bold were novel variants.
All SCN5A variants were missense variants. Of the nine variants, according to ACMG 2015 classification, three variants were classified as likely pathogenic (LP), one variant as pathogenic (P), four variants as variants of uncertain significance (VUS), and one variant as likely benign (LB). Four novel SCN5A gene variants were reported in this study, including p.E901D, p.F853L, p.L377F, and p.H184R. SCN5A variants were distributed along the Nav1.5 amino acid sequence; specifically as follows: one at the C-terminal (p.V1951M); one at the DI transmembrane segment (p.L377F); two at the DII transmembrane segment (p.E901D and p.F853L); and three at intracellular loops (p.R659W, p.R965C, and p.V1525M) (Figure 2). According to in silico prediction tools, eight out of nine SCN5A variants were predicted to be likely pathogenic, with only one variant predicted to be benign (Table 3).
Most minor gene variants were missense variants, with only one synonymous variant (DSC2 p. R136R), one frameshift variant (DSC2 p. L844Dfs*2), and one intron splice site variant (JUP c.1498-5G>C). Among the 27 minor gene variants, according to ACMG 2015 classification, most were classified as variants of uncertain significance (VUS); no variants were classified as pathogenic or likely pathogenic. Five novel variants of minor genes were reported in this study, including two ANK2 variants (p. S2845L and p. V1497L); one CACNA1C variant (p. M1126V); one DSP variant (p. K478N); and one JUP variant (c.1498-5G>C). According to in silico prediction tools, only three out of 27 minor gene variants were predicted to be likely pathogenic, including DSP p. K478N, KCNE2 p. V69M, and CACNA1C p. M1126V. Most other variants were predicted to be benign or of uncertain significance.
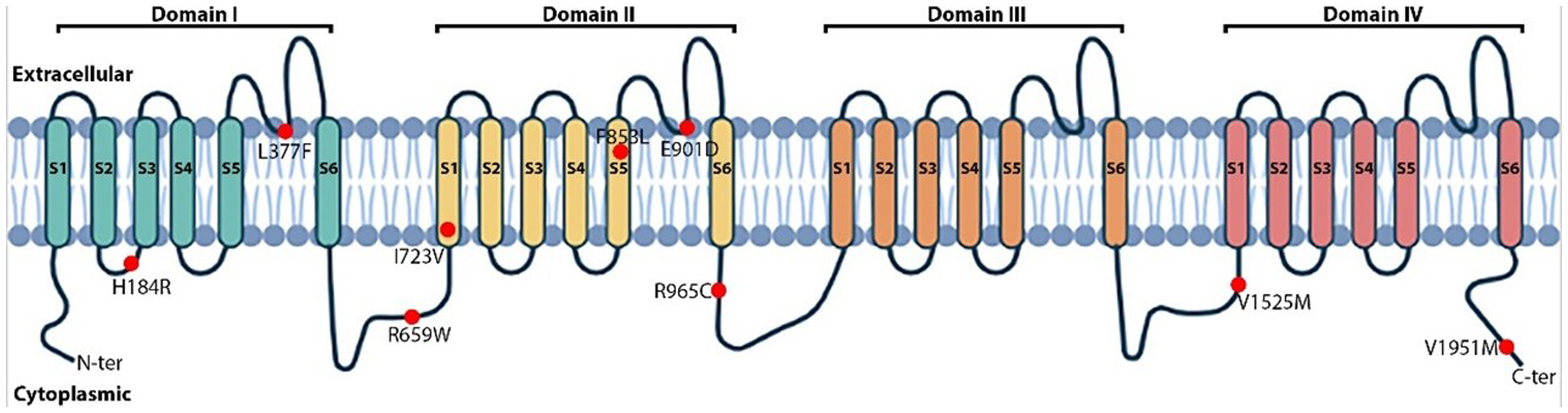
Visual depiction of SCN5A variants in the cardiac voltage-gated sodium channel (Nav1.5).
Discussion
This study was conducted with the primary aim of characterizing the spectrum of genetic background in the Vietnamese BrS population. The majority of variants in our study were SCN5A variants, comprising 25% of the total variants, with an associated prevalence of SCN5A carriers at 16%. In contrast, variants of other minor genes only accounted for less than 10% of the participants. This finding was consistent with previous reports, which indicated that SCN5A is the gene with highest prevalence of BrS-associated variants, ranging from 25% to 30%, while the collective contribution of all other genes accounted for only up to 10% of BrS confirmed cases.24–27 Moreover, our findings implied the pathological significance of SCN5A variants, as 4/9 SCN5A variants in the study were classified as pathogenic or likely pathogenic according to the ACMG standards, while no pathogenic variant was identified in the minor genes. In a 2018 meta-analysis which included 130 studies, the pathogenicity and association with clinical phenotypes of 21 BrS-associated genes were independently reviewed by three gene curation teams. The findings revealed that among variants classified as P/LP, SCN5A variants accounted for up to 94%, whereas variants in the remaining minor genes collectively constituted only 6%. Notably, SCN5A was the sole gene confidently classified as causative for BrS, while other genes were considered disputable by the expert final classification due to insufficient evidence. 10
Pathogenesis of variants according to in silico prediction tools.
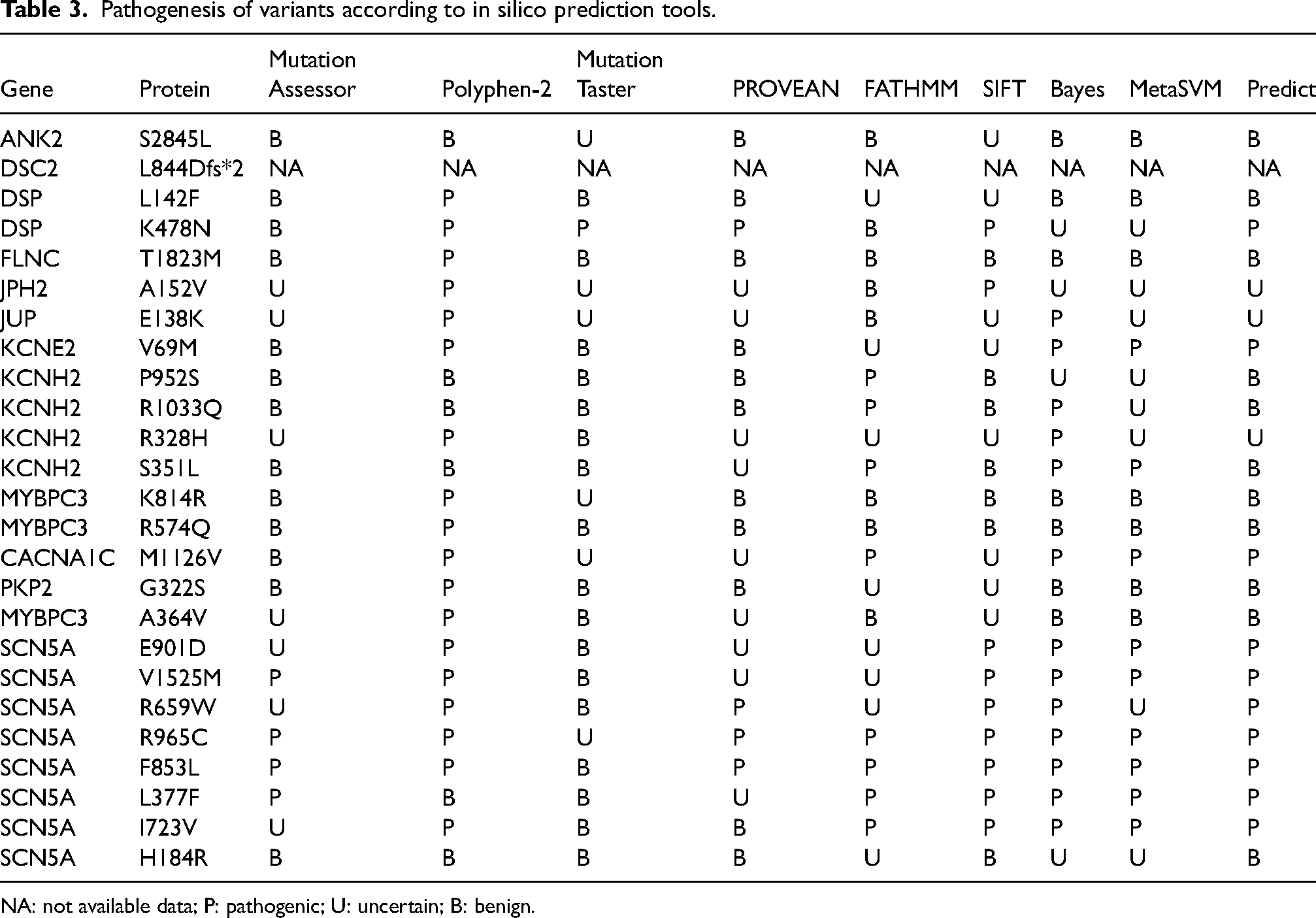
NA: not available data; P: pathogenic; U: uncertain; B: benign.
Our study results demonstrated an association between various clinical phenotypes with SCN5A (+) genotype, including VT/VF induced on EPS, as well as the trends toward higher prevalence of aVR signs, type 1 BrS pattern in peripheral leads, and higher S wave amplitude in D2. Evidence from previous studies did not show a correlation between VT/VF induction during programmed ventricular stimulation and SCN5A variants,28–30 however, the association between aVR signs and the presence of SCN5A variants has been previously reported in several studies.31,32 As of the present time, the Brugada type 1 electrocardiogram pattern is the only recognized electrocardiographic pattern for the diagnosis of BrS, however, the clinical and ECG phenotypes mentioned above have all been reported to be associated with an increased risk of ventricular arrhythmia events in BrS patients.4,15,17,29,33,34 Therefore, the evidence of genotype–phenotype correlation in our study further strengthened the role of these signs in screening for individuals with BrS and their relatives, especially when the Brugada type 1 electrocardiogram pattern is not consistently present, and pharmacological tests are not always available in clinical practice setting in Vietnam.
The findings from our study highlighted the predominant role of the SCN5A gene in BrS, as evidenced by variant prevalence, carrier rates, pathogenesis according to ACMG classification and predictions from in silico tools, as well as its association with clinical phenotypes. In this context, evidence from our study appeared to support the single-gene approach, which aligns with current guidelines, as SCN5A remains the only gene recommended for genetic analysis in BrS patients. Interpretation of all other variants in minor genes should be approached cautiously due to limited evidence regarding their causative role in BrS.
Four novel likely pathogenic missense variants of SCN5A (p.E901D, p.F853L, p.L377F, and p.H184R) were located in S5–S6 segment of domain I and domain II. The extracellular linkers between the S5 and S6 segments constitute the region forming the central pore structure, responsible for controlling the sodium current across the membrane.35,36 Therefore, structural changes in this region, whether minor or major, could lead to the loss or reduction of sodium channel function, contributing to the characteristic ventricular conduction disturbances in BrS. CACNA1C encodes a subunit of a voltage-gated calcium channel, and mutations in this gene have been associated with various cardiac arrhythmias, including BrS. However, CACNA1C mutations are less common and their exact contribution to the syndrome is still being investigated. Studies suggest that alterations in calcium channel function due to CACNA1C mutations may lead to abnormal electrical activity in the heart, contributing to the arrhythmias seen in BrS. 37 ANK2 encodes ankyrin-B, a critical protein involved in the organization and stability of ion channels, particularly sodium channels, within the cell membrane. Mutations in the ANK2 gene are associated with disturbances in ankyrin-B function, leading to dysfunctional sodium channel regulation in cardiac cells. This aberration in sodium channel dynamics can predispose individuals to arrhythmias, such as ventricular tachycardia and ventricular fibrillation, characteristic of BrS. 38 DSP encodes a protein involved in the structure and function of desmosomes, which play a crucial role in maintaining the structural integrity of cardiac tissue. Mutations in the DSP gene can disrupt desmosome function, leading to abnormalities in cardiac cell adhesion and communication. These disruptions can contribute to the characteristic ECG changes and arrhythmias seen in BrS. The exact mechanisms through which DSP mutations lead to BrS may involve complex interactions within the cardiac tissue and ion channel function, and ongoing research is focused on understanding these processes in more detail.39,40 In a 2018 systematic review of 133 rare variants associated with BrS, six ANK2, 12 CACNA1C, and two DSP variants were reported. Four ANK2 variants were classified as LP (c.3382C>G, c.3914G>A, c.7334A>G, and c.10354T>G), two ANK2 variants (c.5758G>A and c.8843C>G) and one DSP variant were categorized as VUS (c.1150G>C), while another DSP variant was classified as LB. 27 The novel variants of minor genes in our study were classified as LB/VUS according to the ACMG classification. However, the predictive outcomes of in silico tools showed considerable inconsistencies. Consequently, additional functional evidence is necessary to confirm the pathogenicity of these variants.
In our study, there were no differences in the time to arrhythmic events between SCN5A carriers/minor gene carriers and the wild-type group. The limitations of our study, including a relatively small sample size and the lack of a follow-up period, may have contributed to the inability to identify such associations.
Conclusion
Our study underscores the primary significance of the SCN5A gene in BrS, as indicated by variant prevalence, carrier rates, pathogenicity per ACMG classification, in silico predictions, and its correlation with clinical phenotypes. This reinforces the single-gene approach advocated by current guidelines, with SCN5A being the sole gene recommended for genetic analysis in BrS patients. Longitudinal study with larger sample size, pedigree, Sanger sequence confirmation, and functional analysis is recommended.
Footnotes
Acknowledgments
We would like to express our sincere gratitude for the valuable insights provided by Mr Dat Thanh Ta throughout the process of ideation, conducting research, and drafting the manuscript for this article. We wish him the best of luck in his journey as a PhD student in Japan and hope for his continued peace and happiness.
Author contributions
Conceptualization: Viet Tuan Tran and Van Khanh Tran; Methodology: Viet Tuan Tran and Van Khanh Tran; Formal analysis and investigation: Viet Tuan Tran; Writing—original draft preparation: Viet Tuan Tran; Writing—review and editing: Viet Tuan Tran, Hung Manh Pham, Phong Dinh Phan, Thinh Huy Tran, and Van Khanh Tran; Resources: Viet Tuan Tran; Supervision: Van Khanh Tran and Hung Manh Pham. All authors read and approved the final manuscript.
Data availability statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.