
Abstract
Aims
Statins are pivotal to the secondary prevention of major adverse cardiovascular events, but some patients are statin-intolerant. We examined the effects of the proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitor alirocumab on the risk of major adverse cardiovascular events according to the intensity of background statin treatment.
Methods and results
The ODYSSEY OUTCOMES trial compared alirocumab with placebo in 18,924 patients with acute coronary syndrome and dyslipidaemia despite intensive or maximum-tolerated statin treatment (including no statin if intolerance was documented). The primary outcome (major adverse cardiovascular events) comprised coronary heart disease death, non-fatal myocardial infarction, ischaemic stroke, or unstable angina. Median follow-up was 2.8 years. Baseline statin treatment was high-intensity (88.8%), low/moderate-intensity (8.7%) or none (2.4%). Median baseline low-density lipoprotein cholesterol was 86, 89 and 139 mg/dL (P < 0.001) in these statin treatment categories, respectively. Alirocumab produced similar relative reductions in low-density lipoprotein cholesterol from baseline across statin treatment subgroups, but the mean absolute reductions differed (52.9, 56.7 and 86.1 mg/dL, respectively; P < 0.001). With placebo, the incidence of major adverse cardiovascular events was highest in the no statin subgroup (10.8%, 10.7% and 26.0% respectively). Alirocumab reduced major adverse cardiovascular events in each statin subgroup (hazard ratio 0.88, 95% confidence interval (CI) 0.80–0.96; 0.68, 0.49–0.94; and 0.65, 0.44–0.97, respectively; Pinteraction = 0.14) with a gradient of absolute risk reduction: 1.25%, 95% CI 0.34–2.16; 3.16%, 0.38–5.94; 7.97%, 0.42–15.51; Pinteraction = 0.106).
Conclusions
PCSK9 inhibition with alirocumab reduces the relative risk of major adverse cardiovascular events after acute coronary syndrome irrespective of background statin treatment. However, patients on no statin are at high absolute risk for recurrent major adverse cardiovascular events; alirocumab substantially reduces that risk. PCSK9 inhibition may be an important therapeutic strategy for statin-intolerant patients with acute coronary syndrome.
Keywords
Background
Statins are a cornerstone of primary and secondary prevention of atherosclerotic cardiovascular events.1,2 Statins are generally well tolerated, with only 2–3% of patients discontinuing treatment due to adverse events in either arm of randomised, placebo-controlled trials. 3 However, this low rate of discontinuation may reflect the selection of patients for clinical trials. In routine clinical practice, inability or unwillingness to continue statin treatment occurs in up to 20% of patients. 3 Similarly, observational studies have shown that a substantial proportion of patients cannot or do not take statins as prescribed due to intolerance, non-adherence, or barriers to accessing medication, with adherence rates ranging from 25% to 60% in different clinical settings. 4 Although many definitions of statin intolerance have been proposed,5,6 a pragmatic operational definition may be the inability to tolerate statin treatment, usually due to the occurrence of symptoms and/or laboratory abnormalities.
High-intensity statin therapy is recommended for most patients with established coronary heart disease 7 or who are required to reach guideline-directed low-density lipoprotein (LDL) cholesterol targets. 8 Because statin intolerance and non-adherence is associated with increased cardiovascular morbidity and mortality,9–11 the potential efficacy of alternative lipid-lowering therapies to improve outcomes in statin-intolerant patients with acute coronary syndromes (ACS) has high clinical relevance.
Proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors reduce levels of LDL-cholesterol by 40−60% in statin-intolerant as well as statin-tolerant patients, but we lack corresponding cardiovascular outcome data in the former group.12–14 Large cardiovascular outcomes trials have shown that PCSK9 inhibitors significantly reduce major adverse cardiovascular events (MACE) in patients with chronic atherosclerotic cardiovascular disease 12 or after a recent ACS when added to background statin treatment. 15 An unanswered question is, therefore, whether the cardiovascular outcomes benefits of PCSK9 inhibitors vary with the intensity of background statin treatment, including no statin treatment in cases of contraindications or statin intolerance. The ODYSSEY OUTCOMES trial compared alirocumab with placebo in patients with a recent ACS and elevated atherogenic lipoproteins despite intensive or maximum-tolerated atorvastatin or rosuvastatin treatment, in some cases no statin.15,16 A determination of statin intolerance (defined per study protocol) was intolerance to at least two statins at any dose. Patients with previous statin intolerance were eligible for enrolment provided intolerance was documented. In this report, we examine the outcomes of treatment with alirocumab or placebo according to the intensity of background statin therapy, including statin intolerance.
Methods
Study design
The design 16 and principal results15,17 of the ODYSSEY OUTCOMES trial (clinicaltrials.gov: NCT01663402) have been reported. In this randomised, multinational, double-blind, placebo-controlled trial, 18,924 patients aged 40 years and over who had been hospitalised with an ACS one to 12 months previously were randomly assigned (1:1) to receive alirocumab or placebo. 16 To be included, patients had to have LDL-cholesterol of 70 mg/dL (1.81 mmol/L) or greater, non-high-density lipoprotein (HDL) cholesterol of 100 mg/dL (2.59 mmol/L) or greater, or apolipoprotein B of 80 mg/dL or greater after 2 or more weeks of stable treatment with atorvastatin 40–80 mg daily, rosuvastatin 20–40 mg daily, or the maximum-tolerated dose of one of these statins (including no statin in the case of documented intolerance). Ethics committees approved the protocol and amendments and all patients provided written informed consent.
Background statin use
If treatment with high-dose atorvastatin or rosuvastatin was not tolerated due to adverse events or laboratory abnormalities (e.g. elevated creatine kinase and/or transaminases), lower doses of atorvastatin (10–20 mg daily) or rosuvastatin (5–10 mg daily) were used, or the patient was switched from atorvastatin to rosuvastatin or vice versa. In the absence of tolerability issues, low/moderate doses of atorvastatin or rosuvastatin could be used for valid medical reasons documented on the case report forms, including advanced age, low body mass, or the interaction of a statin with another required medication.
Statin intolerance was defined per study protocol as intolerance to at least two statins at any dose. A determination of statin intolerance required investigator review of the patient’s medical history, discussion with the patient, their family, and/or the treating physician and documentation on case report forms. Patients with documented statin intolerance could qualify for the trial without any background statin therapy. Treatment with non-statin lipid-lowering drugs was acceptable, with or without concurrent statin therapy, but fibrates other than fenofibrate or fenofibric acid were not acceptable.
Study treatment
Qualifying patients were randomly assigned to alirocumab 75 mg or matching placebo given by subcutaneous injection every 2 weeks. The dose of alirocumab was adjusted under blinded conditions to a target LDL-cholesterol level of 25–50 mg/dL (0.65–1.29 mmol/L) by increasing the dose to 150 mg for LDL-cholesterol levels that remained 50 mg/dL (1.29 mmol/L) or greater or substituting placebo for alirocumab if two consecutive, direct measurements of LDL-cholesterol were less than 15 mg/dL (0.39 mmol/L). After random assignment, investigators were advised to maintain constant background lipid-lowering therapy unless safety or tolerability issues arose. Any changes to background lipid-lowering therapy after random assignment were recorded on a case report form.
Participants and physicians were blinded to the treatment allocation. To protect the blind, all treatment kit boxes had the same look and feel and were labelled with a double-blind label. Details on randomisation procedures are included in the Supplementary material.
Outcome
The primary outcome of the trial was MACE, defined as the composite of death due to coronary heart disease, non-fatal myocardial infarction, fatal or non-fatal ischaemic stroke, or unstable angina requiring hospitalisation.
Statistical analysis
Details on the sample size calculation are included in the Supplementary material. Patients were categorised according to statin dose at random assignment: high-intensity (atorvastatin 40–80 mg or rosuvastatin 20–40 mg daily), low/moderate-intensity (lower doses of atorvastatin or rosuvastatin) or no statin use (with or without non-statin lipid-lowering therapies). Baseline variables are summarised as mean (standard deviation (SD)) or median (quartile 1, quartile 3) according to statin intensity and treatment group. Continuous variables among the statin intensity groups were compared using analysis of variance or quantile regression. Categorical variables were compared using logistic regression. The first change in statin use category from baseline was tabulated using a shift table by treatment group. Blinded adjustment of alirocumab dose was evaluated in each statin subgroup.
In each baseline statin use subgroup, the incidence of MACE over time by assigned treatment was described with Kaplan−Meier curves. Relationships between baseline statin subgroup and the risk of MACE in the placebo group were determined by Cox regression in an unadjusted model and in a model that adjusted for demographic and clinical characteristics (sex, age, geographical region, smoking status, baseline LDL-cholesterol and history of myocardial infarction and coronary artery bypass graft before the index event). The relative risk of MACE between the alirocumab and placebo groups and potential heterogeneity of alirocumab treatment effects by statin subgroup were assessed by a Cox model with a term for the interaction between statin subgroup and treatment group. The absolute risk reduction with alirocumab in each statin subgroup was estimated by absolute differences in observed proportions, and a generalised linear model was used to assess the interaction between statin subgroup and treatment group. Sensitivity analysis on the alirocumab treatment effect was also performed using a time-varying covariate model based on the changing statin use status during the trial. In this analysis, periods with missing statin use data were imputed by carrying forward the previous statin use status.
All analyses were conducted on an intention-to-treat basis, including all patients and events from random assignment to the study end date (11 November 2017). Testing was two-sided with no adjustment for multiple comparisons. Analyses were performed using SAS 9.4.
Results
A total of 18,924 patients underwent random assignment at 1315 centres in 57 countries (see Supplementary Table 1) of whom 9462 were assigned to alirocumab and 9462 to placebo (see Supplementary Figure 1). Patients were randomly assigned between November 2012 and November 2015, except in China where 613 patients were randomly assigned between May 2016 and February 2017.
Consistent with the protocol, most patients (16,811/18,924 (88.8%)) received high-intensity treatment with atorvastatin or rosuvastatin at random assignment; 1653 patients (8.7%) received low/moderate-intensity statin treatment and 460 patients (2.4%) received no background statin treatment. In Asia, 75.1% of patients used high-intensity statin at trial entry compared with 90.7% elsewhere. The median follow-up was 2.8 years (interquartile range 2.3–3.4 years).
Baseline characteristics
Baseline characteristics by statin treatment category are described in Table 1. Patients in the no statin subgroup were older, more likely to be women and reside in North America, and had a greater burden of cardiovascular risk factors, including a family history of premature coronary artery disease and a higher prevalence of hypertension, vascular disease, myocardial infarction, percutaneous coronary intervention, coronary artery bypass graft, or peripheral artery disease. Conversely, patients in the no statin subgroup were less likely to be current smokers. The median (quartile 1, quartile 3) overall baseline LDL-cholesterol concentration was significantly higher (P < 0.001) in the no statin group (139, 115–169 mg/dL) than in the low/moderate-intensity statin group (89, 75–106 mg/dL) or the high-intensity statin group (86, 73–102 mg/dL). Within each statin treatment category, baseline characteristics were similar between those randomly assigned to alirocumab or placebo.
Baseline patient characteristics by statin intolerance and statin intensity.

BMI: body mass index; CABG: coronary artery bypass graft; CAD: coronary artery disease; CHF: congestive heart failure; CRP: C-reactive protein; DBP: diastolic blood pressure; GRF: glomerular filtration rate; HDL: high-density lipoprotein; LDL: low-density lipoprotein; PAD: peripheral artery disease; PCI: percutaneous coronary intervention; Q: quartile; SBP: systolic blood pressure; SD: standard deviation.
Changes in statin dose after random assignment
Among patients in the alirocumab group, 12.2% (1156/9462) had reductions in prescribed statin therapy after random assignment. Supplementary Table 2 shows the first change in prescribed statin therapy after random assignment by treatment group and baseline statin use subgroup. The most common change in statin use in both treatment groups was from high to low/moderate-intensity statin (8.9% (749/8380) and 7.6% (644/8431) in the alirocumab and placebo groups, respectively), followed by a shift from high-intensity statin to no statin (4.0% (335/8380) and 3.7% (315/8431), respectively).
A decrease in the intensity of prescribed statin treatment from its level at random assignment was similar in the alirocumab and placebo groups (12.2% vs. 10.8%, respectively) among those in the low/moderate or high-intensity statin groups, while an increase in statin intensity was less frequent in the alirocumab than the placebo group (0.7% vs. 1.2%, respectively) (see Supplementary Table 2).
Changes in assigned study treatment after random assignment
Supplementary Table 3 shows the numbers of patients in each statin treatment subgroup of the alirocumab and placebo groups who had prematurely discontinued assigned study treatment 4 months and 36 months after random assignment for reasons other than death. Numbers were similar in both treatment groups except for the no statin subgroup at 36 months, when 65 (28.6%) patients in the placebo group versus 30 (12.9%) in the alirocumab group had discontinued study treatment. Supplementary Table 3 also shows the numbers of patients in the alirocumab group on doses of 75 mg or 150 mg, or who had blinded substitution of placebo for alirocumab at 4 and 36 months. As expected, patients in the no statin subgroup were more likely to receive the 150 mg dose of alirocumab, while none had blinded substitution of placebo.
LDL-cholesterol after assignment to treatment with alirocumab or placebo
LDL-cholesterol values at baseline and month 4 according to assigned treatment and change from baseline to month 4 are depicted in Figures 1 and 2, respectively. In the placebo group at month 4 there were minimal changes from baseline in LDL-cholesterol in any of the three statin intensity subgroups. In the alirocumab group at month 4, relative reductions from baseline in LDL-cholesterol were similar in the high-intensity, low/moderate-intensity and no statin subgroups (–57.2%, –59.4% and –58.7%, respectively; P = 0.09) but absolute reductions differed significantly in the three subgroups (–52.9, –56.7 and –86.1 mg/dL, respectively; P < 0.001), consistent with the differences in baseline LDL-cholesterol levels in each subgroup. In the placebo group at month 36, increases from baseline in LDL-cholesterol of 14–15% were observed in the high-intensity and low/moderate-intensity statin subgroups but no changes were seen in the no statin subgroup (see Supplementary Figure 2), probably reflecting shifts in prescribed statin dose (see Supplementary Table 3) or statin adherence. In the alirocumab group at month 36, relative reductions in LDL-cholesterol from baseline were −34.9%, −39.8% and −56.8% in the high-intensity, low/moderate-intensity and no statin subgroups, respectively, reflecting the factors affecting levels in the placebo group, plus patient-driven discontinuation of alirocumab and protocol-specified blinded changes in the dose of alirocumab (see Supplementary Table 3).
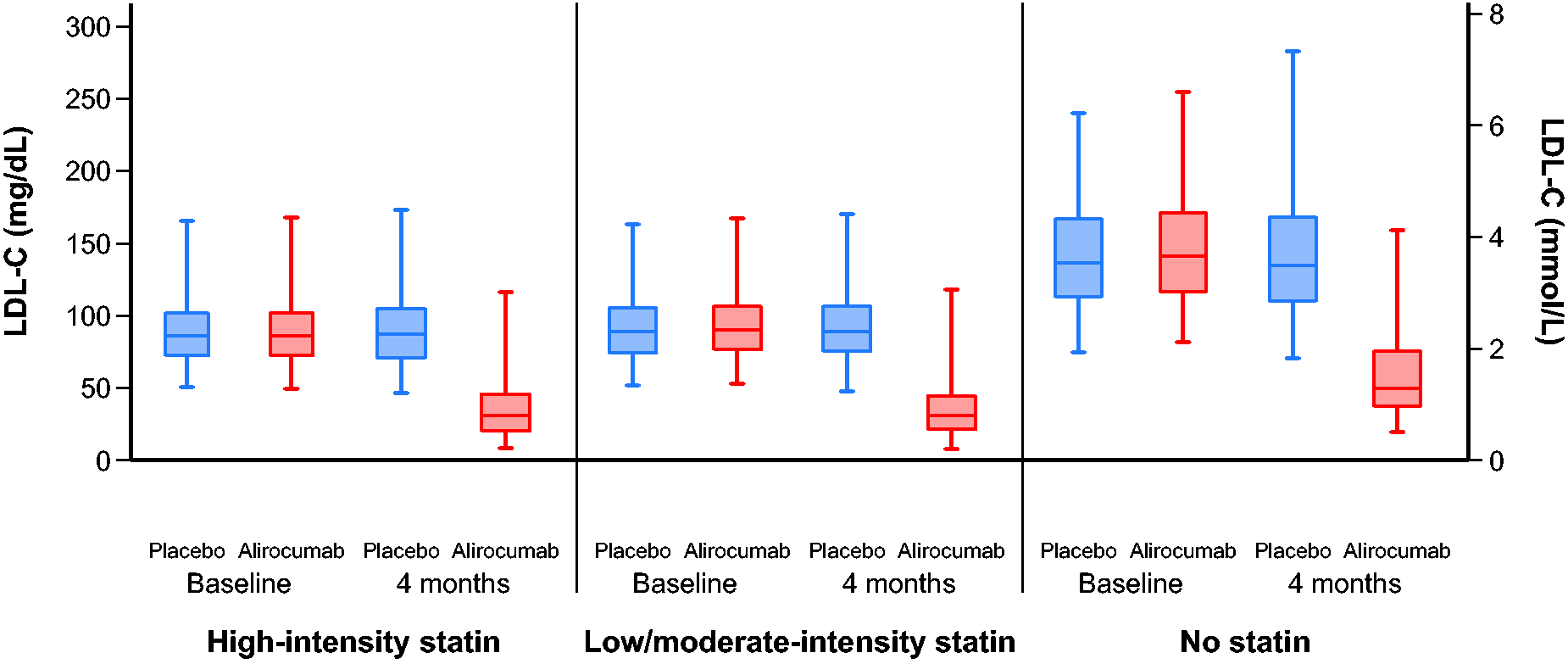
Baseline and month 4 LDL-cholesterol levels by statin intolerance and statin intensity. Lines show medians; boxes, interquartile ranges; whiskers, 2.5th percentile, 97.5th percentile. LDL-C: low-density lipoprotein cholesterol.
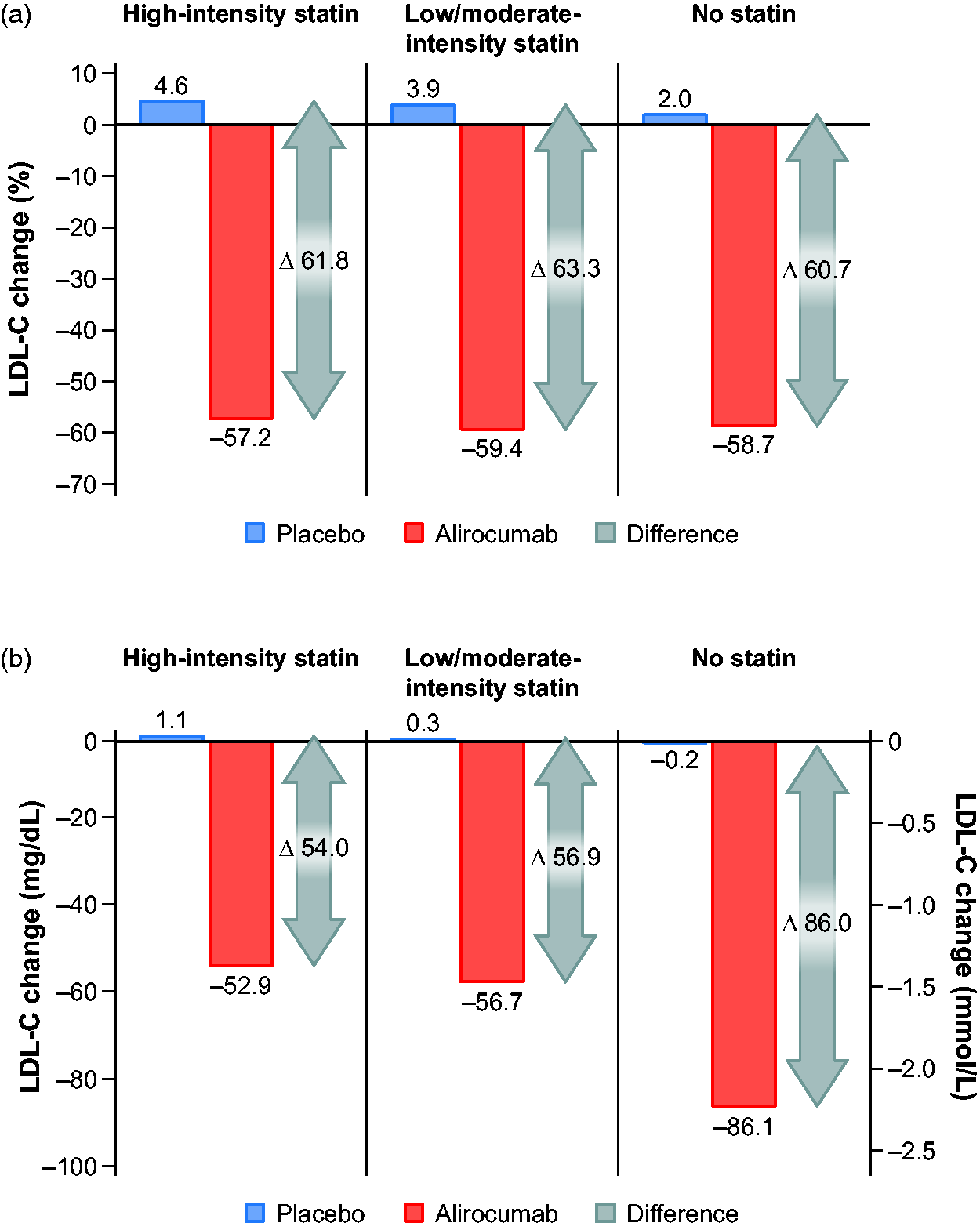
Relative (a) and absolute (b) LDL-cholesterol changes from baseline to month 4 according to statin intolerance and statin intensity. LDL-C: low-density lipoprotein cholesterol.
In the alirocumab group at month 4, 80.6%, 81.9% and 57.6% of patients in the high-intensity, low/moderate-intensity and no statin subgroups achieved the LDL-cholesterol goal of 55 mg/dL (1.4 mmol/L) recommended in current European guidelines. 8 The corresponding percentages in the placebo group were 7.1%, 6.4% and 0.7%.
In the alirocumab group, median (quartile 1, quartile 3) changes in lipoprotein(a) from baseline to month 4 were −5.1 (−13.9, 0), −4.2 (−12.3, 0) and −3.9 (−10.1, 0) mg/dL in the high-intensity, low/moderate-intensity and no statin subgroups, respectively. Corresponding changes in the placebo group were, respectively, 0 (−5.0, 2.6), −0.2 (−4.7, 2.6) and 0 (−2.8, 4.1).
Major adverse cardiovascular events
In the placebo group, the incidence of MACE was markedly higher among those receiving no statin than among those receiving high-intensity or low/moderate-intensity statin treatment, with Kaplan−Meier estimates at 3 years of 29.0%, 11.2% and 11.1%, respectively (Figure 3). Relative to the high-intensity group, the unadjusted hazard ratios (HRs) were 1.08 (95% confidence interval (CI) 0.87–1.35; P = 0.50) and 2.68 (95% CI 2.06–3.50; P < 0.001) for the low/moderate-intensity and no statin groups, respectively. After accounting for imbalances in baseline demographics and clinical characteristics, the risk of MACE was similar between the three groups, with corresponding HRs of 0.97 (95% CI 0.77–1.22; P = 0.79) for the low/moderate-intensity group and 1.10 (95% CI 0.82–1.49; P = 0.52) for the no statin group relative to the high-intensity group.
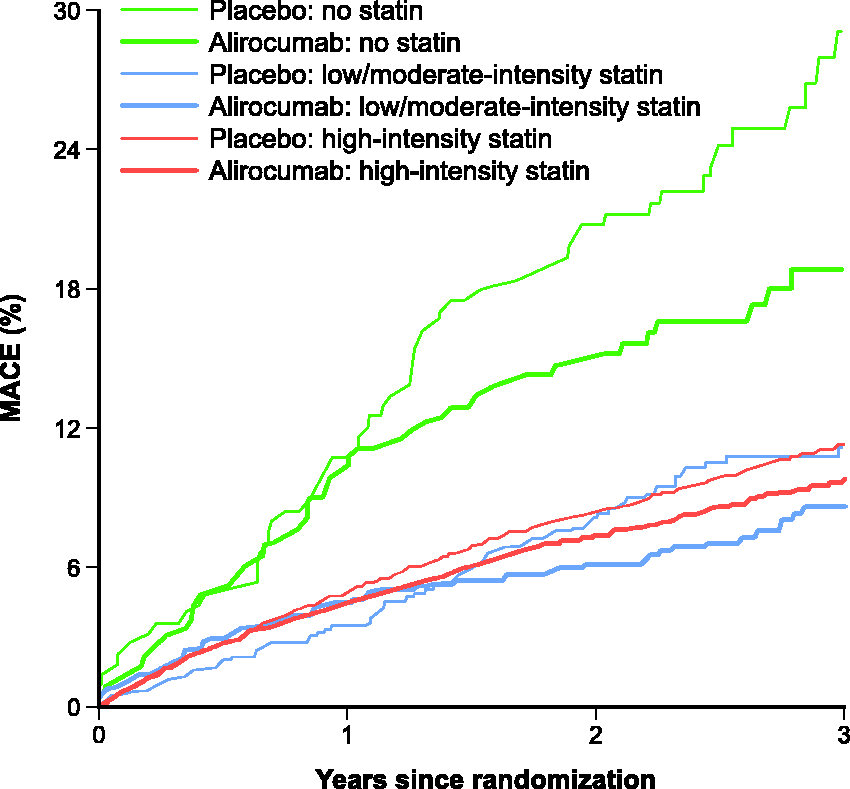
MACE by statin intolerance and statin intensity and treatment group. MACE: major adverse cardiovascular events.
Overall, ODYSSEY OUTCOMES showed a 15% relative risk reduction for MACE with alirocumab versus placebo (HR 0.85, 95% CI 0.78–0.93; P < 0.001). Alirocumab was effective in reducing MACE in each statin subgroup, with numerically smaller HRs in the no statin subgroup (HR 0.65, 95% CI 0.44–0.97) and the low/moderate-intensity subgroup (HR 0.68, 95% CI 0.49–0.94) than in the high-intensity statin subgroup (HR 0.88, 95% CI 0.80–0.96) (Figure 4). The interaction of treatment and statin subgroup on the relative risk of MACE was not significant (P = 0.14). Consistent with the relative risk reduction, the absolute risk reduction with alirocumab in comparison to placebo was larger in the no statin group (7.97%; 95% CI 0.42–15.51) than in the low/moderate-intensity statin group (3.16%; 95% CI 0.38–5.94) and the high-intensity statin group (1.25%; 95% CI 0.34–2.16; Pinteraction = 0.106) corresponding to numbers needed to treat (for 2.8 years) of 13, 32 and 80, respectively.
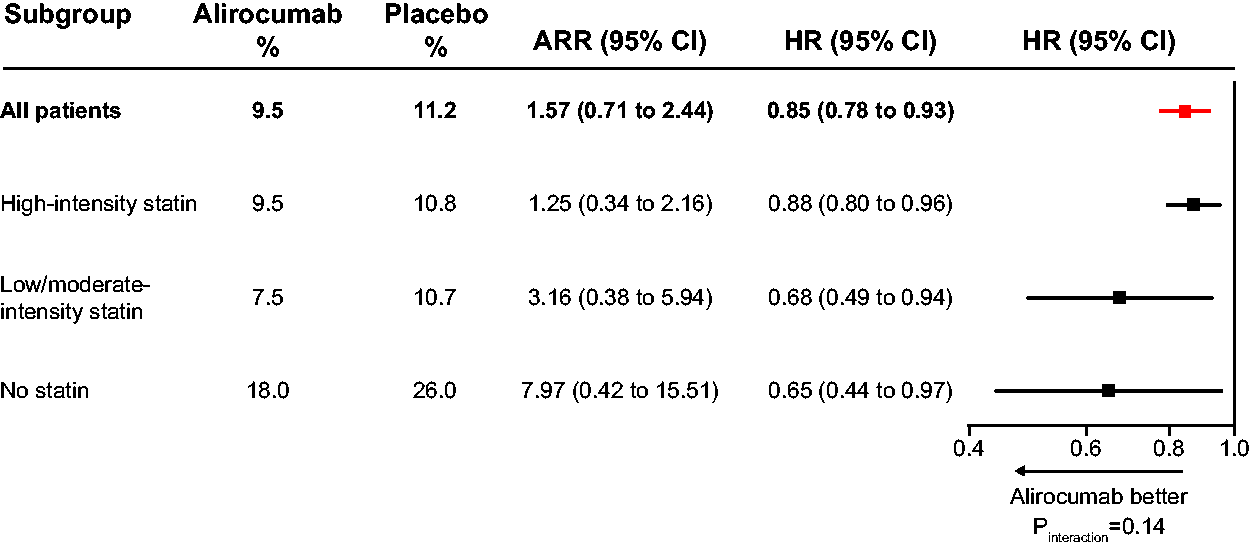
Relative and absolute reductions in MACE by statin intolerance and statin intensity. ARR: absolute risk reduction; CI: confidence interval; HR: hazard ratio; MACE: major adverse cardiovascular events.
In a sensitivity analysis based on time-varying statin use, the HRs for MACE were 0.86 (95% CI 0.78–0.95) for high-intensity statin use, 0.78 (95% CI 0.61–1.01) for low/moderate-intensity statin use and 0.67 (95% CI 0.44–1.01) for no statin use (Pinteraction = 0.400).
Safety and tolerability
Overall, there were minimal differences in the incidence of adverse events or laboratory abnormalities between alirocumab and placebo at all levels of statin intensity, except for local injection-site reactions, which occurred more often in the alirocumab group (see Supplementary Table 4).
Discussion
In the ODYSSEY OUTCOMES trial, patients with a recent ACS who did not receive background statin treatment had high baseline LDL-cholesterol levels, a very high risk of recurrent MACE and large absolute reductions in LDL-cholesterol and MACE with alirocumab. These findings reflect both the absence of statin treatment and the association of no statin treatment with other cardiovascular risk factors. Among patients not treated with statin, the number needed to treat with alirocumab to prevent MACE was fewer than in statin-treated patients. However, the precision of this estimate of absolute benefit was limited by relatively few patients in the no statin subgroup.
Among the 18,924 patients in the trial, 2.4% were not receiving statin therapy at the time of random assignment. This percentage is lower than the prevalence of statin-intolerant patients encountered in clinical practice, most likely because patients are highly motivated to take statin shortly after an ACS and some are statin naive. It may also be because investigators preferentially selected patients able to take statin to participate in the trial, who may be more adherent to prescribed treatments than patients who were not selected.
The relative LDL-cholesterol reductions from baseline to month 4 with alirocumab were approximately 60% in each of the three statin subgroups. However, because baseline LDL-cholesterol concentrations were much higher in the no statin subgroup, the absolute LDL-cholesterol reduction in that subgroup was greater than in the high or low/moderate-intensity statin subgroups.
Similarly, because baseline LDL-cholesterol was higher in the no statin subgroup, fewer patients in that subgroup attained the guideline-recommended 8 LDL-cholesterol target of 55 mg/dL (1.4 mmol/L) compared with patients in the statin-treated subgroups. In the placebo group, almost no patients (0.7%) in the no statin subgroup reached this target; however, with alirocumab, a majority of patients in the no statin subgroup (57.6%) reached it. Thus, treatment with a PCSK9 inhibitor can promote guideline-concordant management of dyslipidaemia in statin-intolerant patients. 18
A discordance between alirocumab-induced changes in LDL-cholesterol and lipoprotein(a) has been reported. 19 In the present analysis, the median changes in lipoprotein(a) from baseline to month 4 were similar in each statin subgroup. Therefore, the larger absolute reduction in the risk of MACE with alirocumab in the no statin subgroup cannot be attributed to a larger reduction in lipoprotein(a) in that subgroup.
Patients in the no statin subgroup were more likely to have protocol-specified uptitration of alirocumab and less likely to have protocol-specified substitution of placebo for alirocumab than patients in the high or low/moderate-intensity statin subgroups, which could have contributed to more pronounced risk reduction with alirocumab in the no statin subgroup.
After random assignment, the most frequent change in prescribed statin treatment was from high-intensity to low/moderate-intensity or no statin treatment. Adherence with prescribed statin was not assessed, but diminishing adherence over time after acute coronary syndrome 20 probably also contributed to the rise in LDL-cholesterol observed in both treatment groups between month 4 and month 36. 21
Among patients assigned to placebo, the incidence of MACE was substantially higher in the no statin subgroup than in the other two statin subgroups, influenced by higher baseline LDL-cholesterol and a greater burden of demographic and clinical comorbidities. Although there was no significant heterogeneity in the relative risk reduction with alirocumab across statin intensity subgroups (Pinteraction = 0.14), the point estimate for the HR was lower in the no statin subgroup (0.65) than in low/moderate-intensity (0.69) or high-intensity (0.88) statin subgroups, in turn contributing to a gradient of absolute risk reduction across the three statin treatment categories. Although the subgroup on no statin treatment comprised a small proportion of the trial cohort, such patients may be more frequently encountered in clinical practice, are readily identifiable and derive a large absolute benefit from alirocumab treatment. However, even in patients receiving and tolerating maximum-intensity statin therapy, alirocumab provided significant clinical benefits.
No trial has specifically focused on the cardiovascular outcomes benefit of alternative lipid-lowering therapies in patients with statin intolerance. However, two trials have shown that PCSK9 inhibitors are well tolerated and produce larger LDL-cholesterol reductions than ezetimibe in such patients.14,22 The GAUSS-3 trial 22 included patients with uncontrolled LDL-cholesterol levels and a history of intolerance to two or more statin. Mean LDL-cholesterol reductions after 24 weeks of treatment were 54.5% with evolocumab and 16.7% with ezetimibe. In ODYSSEY ALTERNATIVE, 14 which compared alirocumab (75 mg every 2 weeks, with a dose increase to 150 mg depending on week 8 LDL-cholesterol value) with ezetimibe in patients at moderate to high cardiovascular risk with statin intolerance, alirocumab reduced LDL-cholesterol by 45.0%, compared with 14.6% with ezetimibe, after 24 weeks of treatment. The CLEAR Tranquility trial 23 compared the effects of bempedoic acid or placebo, added to ezetimibe in statin-intolerant patients. Bempedoic acid reduced LDL-cholesterol by 28.5% more than placebo after 12 weeks of treatment, with similar rates of muscle-related adverse effects to placebo. In summary, these studies indicate that PCSK9 inhibitors, ezetimibe, and other non-statin therapies can reduce LDL-cholesterol concentrations in statin-intolerant patients, albeit with smaller reductions than those achieved with PCSK9 inhibitors. The present analysis adds to these previous data by showing that alirocumab substantially reduces the risk of adverse cardiovascular events in statin-intolerant patients.
Limitations
Several limitations of the current analysis should be noted. First, the ODYSSEY OUTCOMES trial was not specifically designed to determine the efficacy of PCSK9 inhibition in statin-intolerant patients. Second, statin intolerance was defined as patient-reported intolerance to any doses of two statin; it did not require intolerance to the lowest approved doses and was not confirmed with a blinded crossover phase with statin or placebo. Third, we have not analysed data in patients with statin intolerance who received other lipid-lowering therapies because of the small number of such patients. Fourth, patients in the no statin group not only had higher baseline levels of LDL-cholesterol, but also a higher burden of other cardiovascular risk factors. Both probably contributed to the high risk of MACE in the placebo group and the large absolute reduction in MACE with alirocumab.
Conclusions
Intolerance to statin precludes the use of a cornerstone secondary prevention strategy in coronary heart disease and may be particularly relevant to the management of patients with ACS. The present data indicate that statin intolerance is associated with a markedly elevated cardiovascular risk in patients with recent ACS. MACE was reduced regardless of statin intolerance or statin intensity. The availability of lipid-lowering treatment with the PCSK9 inhibitor alirocumab provides an effective therapeutic option for this group of patients to reduce MACE.
Supplemental Material
sj-pdf-1-cpr-10.1177_2047487320941987 - Supplemental material for Intensity of statin treatment after acute coronary syndrome, residual risk, and its modification by alirocumab: insights from the ODYSSEY OUTCOMES trial
Supplemental material, sj-pdf-1-cpr-10.1177_2047487320941987 for Intensity of statin treatment after acute coronary syndrome, residual risk, and its modification by alirocumab: insights from the ODYSSEY OUTCOMES trial by Rafael Diaz, Qian H Li, Deepak L Bhatt, Vera A Bittner, Marie T Baccara-Dinet, Shaun G Goodman, J Wouter Jukema, Takeshi Kimura, Alexander Parkhomenko, Robert Pordy, Željko Reiner, Matthew T Roe, Michael Szarek, Hung-Fat Tse, Harvey D White, Doron Zahger, Andreas M Zeiher, Gregory G Schwartz, Ph Gabriel Steg and for the ODYSSEY OUTCOMES Committees and Investigators in European Journal of Preventive Cardiology
Footnotes
Author contribution
The protocol and statistical analysis plan were conceived by GGS, PGS and MS, developed in conjunction with the other members of the executive steering committee and sponsors, and approved by responsible regulatory authorities and ethics committees. The sponsors participated in study site selection, monitoring and supervision of data collection. Duke Clinical Research Institute led blinded outcome adjudication. An independent data monitoring committee monitored safety and efficacy data. QHL performed the statistical analysis. Analyses were performed independently by the academic statistician in parallel with the sponsors. The manuscript was drafted by RD with input from all authors. RD, DLB, VAB, MTBD, SGG, JWJ, TK, AP, RP, ZR, MTR, MS, HDW, DZ, AMZ, GGS and PGS made substantial contributions to the conception or design of the work; or the acquisition, analysis, or interpretation of data for the work; drafting the work or revising it critically for important intellectual content; and final approval of the version to be published. The executive steering committee decided to publish the paper and takes responsibility for the completeness and accuracy of the data and the fidelity of the trial to the protocol.
Acknowledgments
The author(s) would like to thank the patients, study coordinators and investigators who participated in this trial. Sophie Rushton-Smith and Jenny Lloyd (MedLink Healthcare Communications, London) provided editorial assistance in the preparation of the manuscript (limited to editing for style, referencing and figure and table editing) and were funded by Fondation Assistance Publique–Hôpitaux de Paris, Paris, France.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Dr Diaz reports research grants from Sanofi, DalCor Pharmaceuticals, Population Health Research Institute, Duke Clinical Research Institute, the TIMI group, Amgen, Cirius, Montreal Health Innovations Coordinating Center and Lepetit and personal fees, as a member of the executive steering committee, from Amgen and Cirius; and speaker fees from Eli Lilly. Dr Li is an employee of Regeneron Pharmaceuticals, Inc. Dr Bhatt discloses the following relationships – advisory board: Cardax, Cereno Scientific, Elsevier Practice Update Cardiology, Medscape Cardiology, PhaseBio, Regado Biosciences; board of directors: Boston VA Research Institute, Society of Cardiovascular Patient Care, TobeSoft; Chair: American Heart Association Quality Oversight Committee; data monitoring committees: Baim Institute for Clinical Research (formerly Harvard Clinical Research Institute, for the PORTICO trial, funded by St Jude Medical, now Abbott), Cleveland Clinic (including for the ExCEED trial, funded by Edwards), Duke Clinical Research Institute, Mayo Clinic, Mount Sinai School of Medicine (for the ENVISAGE trial, funded by Daiichi Sankyo), Population Health Research Institute; honoraria: American College of Cardiology (senior associate editor, clinical trials and news, ACC.org; vice-chair, ACC accreditation committee), Baim Institute for Clinical Research (formerly Harvard Clinical Research Institute; RE-DUAL PCI clinical trial steering committee funded by Boehringer Ingelheim; AEGIS-II executive committee funded by CSL Behring), Belvoir Publications (editor in chief, Harvard Heart Letter), Duke Clinical Research Institute (clinical trial steering committees, including for the PRONOUNCE trial, funded by Ferring Pharmaceuticals), HMP Global (editor in chief, Journal of Invasive Cardiology), Journal of the American College of Cardiology (guest editor; associate editor), Medtelligence/ReachMD (CME steering committees), Population Health Research Institute (for the COMPASS operations committee, publications committee, steering committee, and USA national co-leader, funded by Bayer), Slack Publications (chief medical editor, Cardiology Today’s Intervention), Society of Cardiovascular Patient Care (secretary/treasurer), WebMD (CME steering committees); other: Clinical Cardiology (deputy editor), NCDR-ACTION Registry Steering Committee (chair), VA CART Research and Publications committee (chair); research funding: Abbott, Afimmune, Amarin, Amgen, AstraZeneca, Bayer, Boehringer Ingelheim, Bristol-Myers Squibb, Chiesi, CSL Behring, Eisai, Ethicon, Ferring Pharmaceuticals, Forest Laboratories, Fractyl, Idorsia, Ironwood, Ischemix, Lilly, Medtronic, PhaseBio, Pfizer, PLx Pharma, Regeneron, Roche, Sanofi Aventis, Synaptic, the Medicines Company; royalties: Elsevier (editor, Cardiovascular Intervention: A Companion to Braunwald’s Heart Disease); site co-investigator: Biotronik, Boston Scientific, CSI, St Jude Medical (now Abbott), Svelte; trustee: American College of Cardiology; unfunded research: FlowCo, Merck, Novo Nordisk, Takeda. Dr Bittner reports grant support from Sanofi, Astra Zeneca, DalCor, Esperion, Bayer, and Amgen, all paid direct to her institution; personal fees from Sanofi; and a research grant from the Medicines Company. Dr Baccara-Dinet is an employee of and holds shares in Sanofi. Dr Goodman reports research grant support (e.g. steering committee or data monitoring committee) and/or speaker/consulting honoraria (e.g. advisory boards) from: Amgen, AstraZeneca, Bayer, Boehringer Ingelheim, Bristol Myers Squibb, CSL Behring, Daiichi-Sankyo/American Regent, Eli Lilly, Esperion, Ferring Pharmaceuticals, GlaxoSmithKline, HLS Therapeutics, Janssen/Johnson & Johnson, Merck, Novartis, Novo Nordisk A/C, Pfizer, Regeneron, Sanofi, Servier; and salary support from the Heart and Stroke Foundation of Ontario/University of Toronto (Polo) chair, Canadian Heart Research Centre and MD Primer, Canadian VIGOUR Centre, Duke Clinical Research Institute, New York University Clinical Coordinating Centre, and PERFUSE. Dr Jukema reports research grants from The Netherlands Heart Foundation, the Interuniversity Cardiology Institute of The Netherlands and the European Commission Seventh Framework Programme; and research support from Amgen, Astellas, AstraZeneca, Daiichi-Sankyo, Lilly, Merck-Schering-Plough, Pfizer, Roche and Sanofi. Dr Kimura reports research grants from Pfizer, Sanofi, MSD and Bayer; honoraria from Kowa, Sanofi, Pfizer, Asteras-Amgen-Biopharma, MSD, Bayer and AstraZeneca. Dr Parkhomenko reports receiving grants and personal fees from Pfizer, Bayer, Janssen, Sanofi, Amgen, Regeneron, Omthera Pharmaceuticals, Esperion Therapeutics Inc. Dr Pordy is an employee of Regeneron Pharmaceuticals, Inc. Dr Reiner has received honoraria from Sanofi. Dr Roe reports research grant funding from Sanofi-Aventis, Astra Zeneca, Patient Centered Outcomes Research Institute, Ferring Pharmaceuticals, Myokardia, Familial Hypercholesterolemia Foundation and Bayer; and consulting or honoraria from Astra Zeneca, Amgen, Cytokinetics, Eli Lilly, Roche-Genentech, Janssen Pharmaceuticals, Regeneron, Novo Nordisk, Pfizer, Sanofi-Aventis, Signal Path and Elsevier Publishers. Dr Szarek reports serving as a consultant or on advisory boards (or both) for CiVi, Resverlogix, Baxter, Esperion and Regeneron Pharmaceuticals. Dr Tse has received research funding from Abbott, AstraZeneca, Bayer, Boehringer Ingelheim, Boston Scientific, Daiichi-Sankyo, Pfizer, Sanofi-Aventis and St Jude Medical (now Abbott). Dr White reports receiving grant support paid to the institution and fees for serving on a steering committee for the ODYSSEY OUTCOMES trial (Evaluation of Cardiovascular Outcomes After an Acute Coronary Syndrome During Treatment With Alirocumab) from Sanofi-Aventis and Regeneron Pharmaceuticals, for the ACCELERATE study (A Study of Evacetrapib in High-Risk Vascular Disease) from Eli Lilly, for the STRENGTH trial (Outcomes Study to Assess Statin Residual Risk Reduction With EpaNova in High CV Risk Patients With Hypertriglyceridemia) from Omthera Pharmaceuticals, for the SPIRE trial (the Evaluation of Bococizumab (PF-04950615; RN 316) in Reducing the Occurrence of Major Cardiovascular Events in High Risk Subjects) from Pfizer USA, for the HEART-FID study (Randomized Placebo-Controlled Trial of FCM as Treatment for Heart Failure With Iron Deficiency) from American Regent; for the CAMELLIA-TIMI study (a Study to Evaluate the Effect of Long-term Treatment With BELVIQ (Lorcaserin HC) on the Incidence of Major Adverse Cardiovascular Events and Conversion to Type 2 Diabetes Mellitus in Obese and Overweight Subjects With Cardiovascular Disease or Multiple Cardiovascular Risk Factors) from Eisai Inc., for the dal-GenE study (Effect of Dalcetrapib vs Placebo on CV Risk in a Genetically Defined Population With a Recent ACS) from DalCor Pharma UK Inc., for the AEGIS-II study from CSL Behring, for the SCORED trial (Effect of Sotagliflozin on Cardiovascular and Renal Events in Patients With Type 2 Diabetes and Moderate Renal Impairment Who Are at Cardiovascular Risk) and the SOLOIST-WHF trial (Effect of Sotagliflozin on Cardiovascular Events in Patients With Type2 Diabetes Post Worsening Heart Failure) from Sanofi-Aventis Australia Pty Ltd. and for the CLEAR Outcomes Study (Evaluation of Major Cardiovascular Events in Patients With, or at High Risk for, Cardiovascular Disease Who Are Statin Intolerant Treated With Bempedoic Acid (ETC-1002) or Placebo) from Esperion Therapeutics Inc. Dr White was on the Advisory Boards for Acetelion, Sirtex and Genentech, Inc. (an affiliate of F Hoffmann-La Roche Ltd, ‘Roche’; Lytics Post-PCI Advisory Board at European Society of Cardiology) and received lecture fees from AstraZeneca. Dr Zahger serves as national coordinator for the ODYSSEY OUTCOMES trial and the SCORED trial, both funded by Sanofi. He has consulted for Bayer, Astra Zeneca, Boehringer-Ingelheim, NovoNordisk and Sanofi. Dr Zeiher reports receiving fees for serving on a steering committee for the ODYSSEY OUTCOMES trial from Sanofi, and advisory board and speaker fees from Sanofi, Amgen, Boehringer Ingelheim, Bayer, Novartis, Pfizer, AstraZeneca and Vifor. Dr Schwartz reports research grants to the University of Colorado from Resverlogix, Roche, Sanofi and the Medicines Company. In addition, Dr Schwartz is co-inventor of pending US patent 62/806313 ‘Methods for Reducing Cardiovascular Risk’, assigned in full to University of Colorado. Dr Steg reports grants and non-financial support (co-chair of the ODYSSEY OUTCOMES trial; as such he received no personal fees, but his institution has received funding for the time he has devoted to trial coordination, and he has received support for some travel related to trial meetings) from Sanofi; research grants and personal fees from Bayer (steering committee MARINER, grant for epidemiological study), Merck (speaker fees, grant for epidemiological studies), Sanofi (co-chair of the ODYSSEY OUTCOMES trial; co-chair of the SCORED trial; consulting, speaking), Servier (chair of the CLARIFY registry; grant for epidemiological research) and Amarin (executive steering committee the REDUCE-IT trial (Disease Reduction of Cardiovascular Events With Icosapent Ethyl–Intervention Trial); consulting); and personal fees from Amgen, Bristol-Myers Squibb, Boehringer Ingelheim, Pfizer, Novartis, Regeneron Pharmaceuticals, Lilly and AstraZeneca. Dr Steg also has a European application number/patent number, issued on 26 October 2016 (no. 15712241.7), for a method for reducing cardiovascular risk.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The trial was funded by Sanofi and Regeneron Pharmaceuticals, Inc. The sponsors participated in the selection of the trial sites, the monitoring of the trial and the supervision of data collection.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.